- Chebbi Amel #amel dot chenaoui at yahoo dot frService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
- Ben Sassi SamiaService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
- Zeghal ImenService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
- Malek InesService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
- Bouguila HediService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
- Nacef LeilaService A. Institut Hédi Raies d’Ophtalmologie. Tunis, Tunisie
Introduction : Les myopathies mitochondriales sont des pathologies héréditaires rares qui affectent les fonctions énergétiques de la mitochondrie. Leur expression clinique est polymorphe et parfois multi-systémique. L’ophtalmoplégie externe chronique progressive en constitue la forme clinique la plus fréquente.
Observation : Nous rapportons l’observation d’un homme de 48 ans atteint d’ophtalmoplégie externe chronique progressive dans le cadre d’une myopathie mitochondriale avec ptosis myogène acquis bilatéral. Un bilan étiologique complet, une biopsie musculaire et une analyse génétique ont permis de conclure à une myopathie mitochondriale. Nous discutons dans ce travail l’approche diagnostique et thérapeutique.
Conclusion : L’ophtalmoplégie externe chronique progressive dans le cadre d’une myopathie mitochondriale est une maladie génétique rare qui pose un problème de prise en charge diagnostique et dont l’approche chirurgicale est décevante vues les récidives fréquentes. L’emploi d’activateurs énergiques qui améliorent la synthèse de l’ATP par les mitochondries dans l’avenir semble prometteur.
Introduction: Mitochondrial myopathy is a rare hereditary disease which affects mitochondrial functions. It appears to have a polymorphic and multisystemic clinical expression. Chronic progressive external ophtalmoplegia is the most frequent clinical form.
Observation: We report the case of a man 48 years old who present a chronic progressive ophtalmoplegia due to mitochondrial myopathy with an acquired bilateral myogenic ptosis.
We performed ophthalmological and neurological examinations, laboratory testing, skeletal muscle biopsy, including histological and histochemical investigations, and molecular genetic testing of skeletal muscle DNA. These investigations proved the diagnosis of mitochondrial chronic progressive external ophthalmoplegia. We discuss in this case the diagnostic and the therapeutic approach.
Conclusion: Mitochondrial chronic progressive external ophthalmoplegia is a rare genetic affection which needs appropriate diagnostic and therapeutic strategies. Energetic activators which improve the ATP synthesis would be an efficient alternative in the future.
Les myopathies mitochondriales constituent un ensemble de maladies génétiques rares regroupant des affections métaboliques où il existe un déficit énergétique de la mitochondrie. D’expression très polymorphe, ces affections à caractère souvent multiviscéral se présentent le plus généralement sous une forme myopathique ou encéphalomyopathique.
La forme la plus courante est l’ophtalmoplégie externe chronique progressive (OECP) qui est une myopathie progressant lentement et intéressant principalement et exclusivement les muscles oculaires externes. Les premiers muscles à être touchés sont les releveurs de la paupière supérieure avec un ptôsis symétrique non fluctuant, généralement sans diplopie, qui sera suivi d’une ophtalmoplégie progressive.
Nous rapportons l’observation d’un homme de 48 ans, qui a rapporté l’installation progressive depuis 20 ans de :
- Troubles oculaires à type de ptôsis bilatéral, de difficulté à la mobilisation des globes oculaires et de larmoiement,
- Troubles phonatoires : la voix était nasonnée avec dysphonie,
- Troubles de la déglutition avec fausses routes fréquentes,
- Fatigabilité précoce lors des efforts.
A l’examen général, on a trouvé :
- Un patient conscient, bien orienté dans le temps et l’espace avec un faciès inexpressif,
- Une voix nasonnée et des troubles de la déglutition,
- Pas de déficit moteur au niveau des ceintures scapulo-humérale et pelvienne,
- Le reste de l’examen neurologique et général sans particularité.
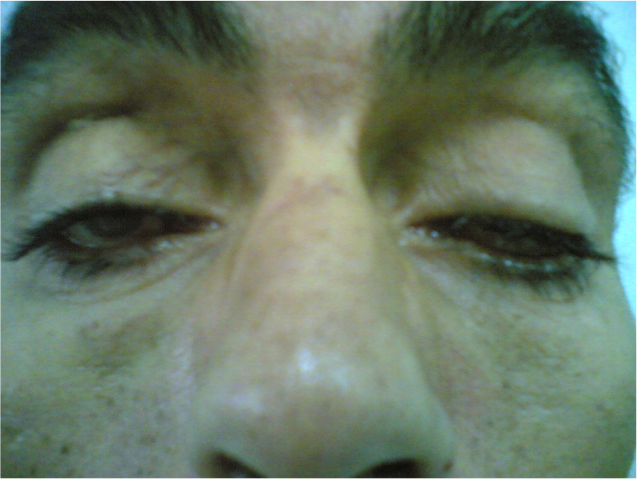
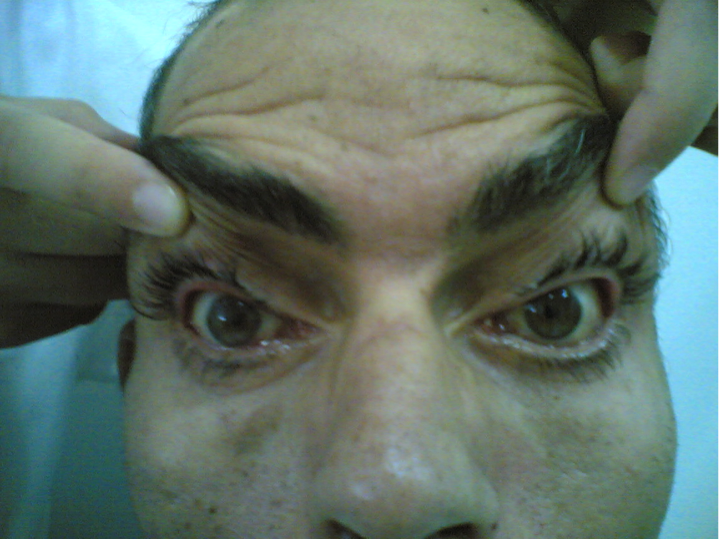
L’examen ophtalmologique a mis en évidence un ptôsis bilatéral symétrique et permanent, en partie compensé par le rejet de la tête en arrière et la contraction des muscles frontaux avec élévation des sourcils (figure 1). Une ophtalmoplégie complète et bilatérale sans diplopie, réalisait le faciès de Hutchinson (figure 2). Les pupilles étaient épargnées avec un réflexe photomoteur présent et symétrique. Il existait également une kératite d’exposition bilatérale avec dégénérescence cornéenne à 6 heures (figure 3). Il n’y avait pas de phénomène de Bell (pas de bascule du globe oculaire en haut et en dedans lors de la fermeture contrariée, bilatérale et symétrique). L’examen du fond d’œil n’a pas révélé d’anomalie, particulièrement il n’y avait pas de rétinite pigmentaire.
Un bilan étiologique complet a été pratiqué.
- Le test à la prostigmine a été négatif et l’EMG n’a pas mis en évidence de bloc de la jonction neuro-musculaire de type myasthénique.
- La biopsie musculaire du deltoïde avait conclu à une myopathie mitochondriale avec participation lipidique. Sur le trichrome de Grimori, il existait une discrète inégalité de la taille des fibres, avec présence de nombreuses fibres denses riches en mitochondries. L’aspect de « ragged red fibers» (fibres rouges déchiquetées) a été retrouvé au niveau d’une dizaine de fibres. Sur les activités oxydatives, ces fibres étaient très riches en mitochondries surtout sur la succinodéshydrogénase. Sur les activités ATPasiques, il existait une répartition égale des fibres I et II avec une tendance au grouping. Sur le noir Soudan, il y avait des fibres très riches en lipides.
Une évaluation cardiologique n’a pas mis en évidence d’autre anomalie.
Le diagnostic d’une ophtalmoplégie externe chronique progressive dans le cadre d’une myopathie mitochondriale a été retenu.

Les myopathies mitochondriales sont des affections liées à un dysfonctionnement de la chaîne respiratoire mitochondriale et caractérisées par la présence dans le muscle d’anomalies morphologiques des mitochondries (fibres rouges déchiquetées). D’expression clinique très polymorphe, ces affections à caractère souvent multiviscéral se présentent le plus généralement sous une forme myopathique (ophtalmoplégie externe chronique progressive, faiblesse musculaire) ou encéphalomyopathique (syndrome de Leigh, MELAS, MERFF…) [1].
Chez l’adulte, l’ophtalmoplégie externe chronique progressive avec ptosis bilatéral est la présentation la plus fréquente des cytopathies mitochondriales. Elle peut être isolée ou compliquée d’une myopathie des ceintures ou d’une atteinte du système neurologique et/ou neurosensoriel : (ataxie, syndrome pyramidal, ralentissement intellectuel, surdité…etc). Enfin, elle constitue avec la rétinite pigmentaire et le bloc auriculo-ventriculaire, le profil clinique typique du syndrome de Kearns-Sayre [1].
Les myopathies oculaires mitochondriales, d’expression clinique pure ou combinée, comportent d’une part un ptosis et une ophtalmoplégie, d’autre part des anomalies mitochondriales sur la biopsie musculaire [1] [2].
Le ptosis, souvent bilatéral est parfois asymétrique et relativement permanent. II est en partie compensé par le rejet de la tête en arrière et la contraction des muscles frontaux avec élévation des sourcils. L’ophtalmoplégie, d’installation très progressive, ce qui explique l’absence de diplopie, entraîne d’abord une diminution de l’élévation du regard puis une paralysie oculomotrice complète qui réalise le faciès de Hutchinson avec immobilité des yeux en position légèrement divergente. Les pupilles sont habituellement épargnées [2].
Le second élément est représenté par les anomalies mitochondriales visibles préférentiellement sur le muscle deltoïde. En microscopie optique, l’image caractéristique est celle des fibres « rouges déchiquetées » (ragged red fibres ou RRF) visible sur les colorations au trichrome de Grimori. La périphérie de la fibre est alors colorée en rouge, de même que la région intermyofibrillaire. Elle est intensément réactive aux colorations oxydatives principalement la succinodéshydrogénase. Elle est également positive avec la cytochrome oxydase mais selon une répartition segmentaire comme le montrent les sections longitudinales. Ces anomalies siègent surtout sur les fibres de type I, plus rarement sur les fibres de type II. Elles sont réparties au hasard sur la biopsie et portent sur environ 5 à 20 % des fibres. En microscopie électronique, des amas de mitochondries anormalement grandes sont visibles sous la membrane plasmique et dans les espaces intermyofibrillaires. Leur taille et leur forme sont variables. Elles contiennent des crêtes allongées ou concentriques et des inclusions paracristallines, souvent rectangulaires, disposées en galons. Des gouttelettes lipidiques en grand nombre et des amas de glycogène sont fréquentes. Ces anomalies mitochondriales ne siègent pas seulement dans le muscle, mais sont susceptibles de s’observer dans divers organes notamment le coeur, le foie et le cervelet. Les cas autopsiés comportent une dégénérescence spongiforme de la substance blanche de l’encéphale et des noyaux du moteur oculaire commun analogue aux encéphalopathies spongiformes [2].
Un certain nombre de myopathies mitochondriales sont rattachées à un remaniement du génome mitochondrial : mutations ponctuelles, délétions ou duplications. L’ophtalmoplégie externe chronique progressive est en relation avec une délétion de l’ADN mitochondrial (ADNmt) [1] [2].
La transmission des myopathies mitochondriales se fait selon différents modes. Dans la mesure où la structure protéique des complexes enzymatiques de la chaîne respiratoire est codée à la fois par l’ADNmt et par l’ADN nucléaire, les différentes formes de déficits peuvent être rattachées à des anomalies du génome mitochondrial et/ou du génome nucléaire [1].
L’hérédité peut donc être maternelle (mutation de l’ADNmt pré-existante chez la mère et transmission possible à toute la descendance), ou mendélienne (autosomale dominante ou récessive). Cependant, la plupart des myopathies mitochondriales se présentent comme des affections sporadiques (mutations intervenant très précocement durant les premiers stades de l’embryogenèse avant l’organogenèse [1].
L’indication opératoire n’est posée que lorsque le ptosis compromet l’axe visuel des deux yeux et ceci notamment à cause des complications secondaires aux problèmes d’exposition dus à la lagophtalmie chez les patients ayant un phénomène de Bell inadéquat ou absent [3] [4].
Divers auteurs recommandent la prudence sur la correction chirurgicale chez ces patients à cause d’une augmentation du risque d’une hypercorrection postopératoire [4] [5].
La technique opératoire utilisée est soit la résection par voie antérieure du releveur de la paupière soit la suspension au muscle frontal sur la base de la fonction résiduelle du releveur.
Certains auteurs tels Savino [5] indiquent une résection par voie antérieure du releveur de la paupière et du muscle de Müller pour les cas présentant une fonction du releveur d’au moins 4-5 mm. La suspension au muscle frontal selon la technique de Fox étant indiquée si la fonction du releveur est inférieure à 4-5 mm. Wong et al [3] indiquent la suspension au muscle frontal que si la fonction du releveur est inférieure à 8 mm. Ceux qui ont une fonction du releveur d’au moins 8 mm sont traités par une résection du releveur.
Les résultats fonctionnels dépendent des problèmes de récidive du ptosis qui est variable selon les auteurs [6] [7].
L’ophtalmoplégie externe chronique progressive dans le cadre d’une myopathie mitochondriale est une maladie génétique rare qui pose le problème de sa prise en charge sur le plan diagnostique et thérapeutique.
De nombreuses tentatives de traitements spécifiques ont été réalisées et semblent prometteuses. Leur principe est de contourner le blocage métabolique en administrant des accepteurs d’électrons se substituant aux complexes déficitaires (vitamine C, K3, B2) ou les transporteurs eux-mêmes (Coenzyme Q10).
Conflits d’intérêts : Aucun
- Mousson B, Maire I, Carrier H, Flocard F, Flechaire A, Vidailhet M. [Mitochondrial cytopathies]. Rev Med Interne. 1991;12:219-26 pubmed
- Serratrice G. In : Ptosis. EMC-Neurologie 2005, 2 :133–47.
- Wong V, Beckingsale P, Oley C, Sullivan T. Management of myogenic ptosis. Ophthalmology. 2002;109:1023-31 pubmed
- Burnstine M, Putterman A. Management of myopathic ptosis. Ophthalmology. 2004;111:411; author reply 411 pubmed
- Savino G, Di Nicola D, Quaranta-Leoni F, Dickmann A. [Surgical treatment of ptosis in cases of mitochondrial myopathies and severe myasthenia]. J Fr Ophtalmol. 1994;17:4-9 pubmed
- Patte M, Dalens H, Sole P, Sole M, Clavelou P, Boespflug-Tangy O, et al. [A familial case of chronic progressive external ophthalmoplegia associated with mitochondrial disease]. J Fr Ophtalmol. 2001;24:961-5 pubmed