Introduction : Le sarcome alvéolaire des parties molles (SAPM) est une tumeur maligne rare, d’étiologie inconnue touchant avec prédilection l’adolescent et l’adulte jeune. Il possède des caractéristiques cliniques, histologiques, moléculaires et thérapeutiques distinctes. Observation : Il s’agissait d’un patient âgé de 19 ans, sans antécédent pathologique, qui avait présenté une tuméfaction douloureuse du périnée. L’imagerie avait objectivé une masse de la fosse ischio-rectale gauche refoulant le rectum et la loge prostatique sans envahissement tumoral. Une biopsie de la masse a mis en évidence une prolifération tumorale faite de cellules de grande taille, polygonales agencées en lobules de taille variable séparés par des fins septas fibro-vasculaires. Le complément immuno-histochimique à l’aide d’anticorps anti TFE3 confirmait le diagnostic du sarcome alvéolaire des parties molles. Le patient a eu une exérèse de la masse tumorale. L’évolution était marquée par l’apparition de métastases pulmonaires. Le patient est mis sous chimiothérapie. Conclusion : Le sarcome alvéolaire des parties molles est caractérisé par une translocation quasi spécifique t(X;17)(p11;25) qui crée une protéine de fusion ASPL-TFE3 agissant comme facteur de transcription aberrant. L’évolution est souvent indolente malgré un fort potentiel métastatique.
Introduction: Alveolar soft tissue sarcoma is a rare malignancy of unknown etiology with a predilection for adolescents and young adults. It has distinct clinical, histological, molecular and therapeutic features. Case: A 19-years-old patient, with no medical history, presented painful swelling of the perineum. The radiology revealed a mass of the left ischio-rectal fossa pushing back the rectum and the prostatic lodge without tumor invasion. A mass biopsy showed a tumor proliferation made of large, polygonal cells arranged in lobules of different sizes separated by fibro-vascular septa. Immunohistochemical analysis with anti-TFE3 antibodies confirmed the diagnosis of alveolar soft tissue sarcoma. The patient underwent an excision of the tumor mass. The outcome was marked by the appearance of pulmonary metastases. The patient received chemotherapy. Conclusion: Alveolar soft tissue sarcoma is characterized by an almost specific translocation t(X, 17)(p11;25) which creates a fusion protein, APSL-TFE3, acting as an aberrant transcription factor. The evolution is often indolent, with long-term survival despite a high rate of metastases.
Le sarcome alvéolaire des parties molles (SAPM) est une tumeur maligne rare de localisation préférentielle au niveau des membres et survenant généralement chez les adolescents et les adultes jeunes [1]. C'est une tumeur possédant des caractéristiques spécifiques cliniques, histologiques, moléculaires et thérapeutiques. L’évolution est souvent indolente, avec une survie à long terme malgré un taux élevé de métastases [2]. Nous rapportons un cas rare de SAPM de localisation périnéale et à travers lequel nous discuterons les différentes données épidémiologiques, cliniques, histopathologiques et moléculaires disponibles dans la littérature.
Un jeune patient âgé de 19 ans, sans antécédent pathologique, présentait depuis 6 mois une tuméfaction douloureuse du périnée avec augmentation progressive du volume sans signes digestifs ni urinaires associés. Le tout évoluait dans un contexte d’apyrexie et d’altération progressive de l’état général. Le bilan radiologique (Tomodensitométrie et Imagerie par résonnance magnétique IRM) a mis en évidence une masse tumorale de la fosse ischio-rectale gauche refoulant le rectum et la loge prostatique sans envahissement tumoral (Figure 1).

Une biopsie exérèse de la masse a été réalisée. Histologiquement, il s’agissait d’une prolifération tumorale faite de lobules de taille variable séparés par des fins septas fibro-vasculaires. Les cellules tumorales sont de grande taille, polygonales, pourvues de cytoplasme abondant clair parfois éosinophile. Les noyaux sont de taille variable, vésiculeux renfermant un nucléole central proéminent. L’activité mitotique est faible (moins de 2 mitoses par 10 champs au fort grossissement). Ces cellules tumorales se détachent vers le centre des lobules tumoraux (Figure 2).
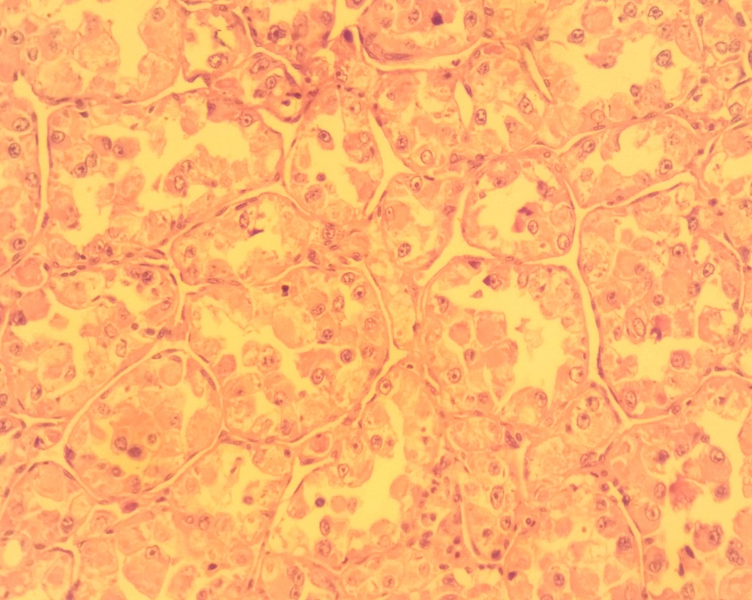
Des images d’embols vasculaires sont observées à la périphérie de la tumeur (Figure 3).

Devant cet aspect morphologique, une métastase d’un carcinome à cellules rénales, un paragangliome, un mélanome achromique ou un rhabdomyosarcome alvéolaire ont été évoqués. À l’examen immuno-histochimique, les cellules tumorales n’exprimaient pas les antigènes suivants : cytokératine AE1-AE3, vimentine, CD10, chromogranine, synaptophysine, MélanA, HMB45, PS100, Actine muscle lisse et la myogénine. Un 2ème panel a été réalisé à l’aide d’anticorps anti TFE3 s’est révélé positif (Figure 4).
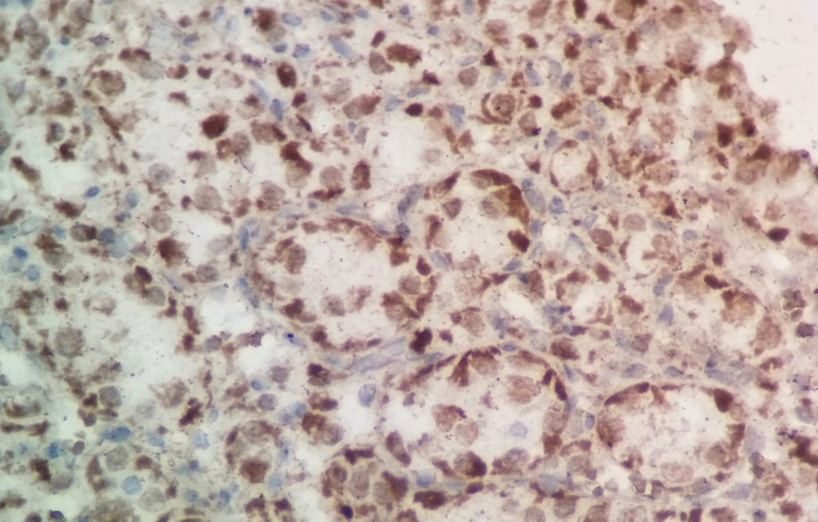
L’aspect histopathologique et immuno-histochimique était compatible avec le diagnostic d’un sarcome alvéolaire des parties molles. Le patient a été opéré avec exérèse de la masse tumorale. L’évolution était marquée par l’apparition de métastases pulmonaires. Le patient est mis sous chimiothérapie adjuvante.
Le sarcome alvéolaire des parties molles (SAPM) est une tumeur maligne rare, d’étiologie inconnue, appartenant, selon la classification 2013 de l’Organisation Mondiale de la Santé, au groupe des tumeurs de différenciation incertaine [3]. Il a été décrit et caractérisé pour la première fois en 1952 par un pathologiste, le docteur W. Christopherson [4]. Il posséde des caractéristiques spécifiques cliniques, histologiques, moléculaires et thérapeutiques [5]. C’est un sarcome rare représentant 0,2 à 0,9 % de tous les sarcomes des tissus mous. Il affecte surtout l'adolescent ou l'adulte jeune de 15 à 35 ans. Il est rare avant 5 ans et après 50 ans. Il existe une légère prédominance féminine chez l'enfant et l'adulte jeune avant l'âge de 30 ans (sex ratio : 2). Chez l’adulte, la localisation la plus commune est représentée par les tissus mous profonds des cuisses et des fesses. Chez l’enfant, c’est la région de tête et cou, et de manière très particulière, la langue et l'orbite. Des cas isolés ont été rapportés au niveau du poumon, foie, estomac, os, cœur, vessie, etc. Sur le plan clinique, le SAPM se présente comme une masse profonde à croissance lente et non douloureuse [3]. Le délai entre les premiers symptômes et le diagnostic est compris entre 1 à 7 mois. Les métastases sont fréquemment présentes au moment du diagnostic et sont retrouvées dans près d'un tiers des cas. Les sites préférentiels de celles-ci sont les poumons, puis les os et le système nerveux central. L'existence de métastases ganglionnaires est par contre exceptionnel (moins de 5 %) [4]. Compte tenu de la rareté de cette pathologie, il y a peu de données radiologiques spécifiques. L’hypervascularisation tumorale avec un drainage veineux proéminent peut être mise en évidence par angiographie ou par IRM avec injection de Gadolinium et permet d’évoquer le diagnostic de SAPM [3]. Sur le plan macroscopique, Il s'agit d'une masse profonde de plusieurs centimètres mal limitée située dans le muscle squelettique ou les tissus mous profonds, de couleur jaune ou grise pâle. Les foyers nécrotiques et hémorragiques sont surtout observés dans les tumeurs volumineuses. Sur le plan histologique, l'aspect est le plus souvent caractéristique, le terme « alvéolaire » se rapporte à l'architecture en nids des cellules tumorales avec au centre un espace vide et en périphérie un fin réseau vasculaire, rappelant l'architecture alvéolaire du poumon. Cet aspect peut être secondaire à une perte de la cohésion cellulaire et la nécrose au centre des lobules [3]. Ces nids sont organisés en compartiments et en lobules séparés par des septas fibreux. Les cellules tumorales sont de grande taille, uniformes, au cytoplasme abondant, polygonal éosinophile et granuleux, au noyau rond, excentré contenant un nucléole unique de grande taille. Les mitoses et la nécrose sont rares. Le cytoplasme contient des cristaux caractéristiques, rhomboïdes ou polygonaux PAS positifs et résistant à la diastase dans 80 % des cas. Une infiltration vasculaire est souvent présente à la périphérie de la tumeur [4]. Il existe quelques variantes histologiques, l'aspect des cellules tumorales peut faire discuter d'autres diagnostics: rhabdomyosarcome alvéolaire, sarcome épithélioïde, tumeur à cellules granuleuses, hibernome, paragangliome, rhabdomyome, mélanome, corticosurrénalome, carcinome du rein, hépatoblastome, PECome (perivascular epithelioid cell) mais le contexte clinique, les données morphologiques et l'immunophénotypage sont différents. Sur le plan immunophénotypique, les cellules tumorales montrent de façon caractéristique une surexpression nucléaire du Transcription factor for immunoglobulin heavy-chain Enhancer 3 (TFE3). Cette sur-expression de TFE3 n'est pas spécifique car elle s'observe aussi dans les carcinomes du rein avec translocation impliquant TFE3, dans certaines tumeurs à cellules granuleuses et dans certains PEComes [4]. Les cellules tumorales peuvent exprimer la desmine et la protéine S100 de façon focale et faible. Elles n’expriment pas la myogénine, HMB45, kératines, chromogranine et la synaptophysine. Le diagnostic différentiel du SAPM se pose essentiellement avec une métastase d'un carcinome rénal; mais il s'agit en général de malades plus âgés, le bilan radiologique permet de retrouver la tumeur rénale initiale, la coloration au PAS ne montre pas de structure cristalline et l'immunomarquage permet d'identifier la nature épithéliale de la tumeur. Le paragangliome montre une topographie différente et un profil immunohistochimique particulier (positivité de PS100 (cellules sustentaculaires), synaptophysine et chromogranine) et plus rarement une tumeur à cellules granuleuses (protéine S100 positive), un angiosarcome, un mélanome malin achromique, un rhabdomyosarcome alvéolaire ou un PECome [6]. Sur le plan génétique, le sarcome alvéolaire des parties molles est caractérisé par une translocation spécifique, souvent non réciproque, t(X;17) (p11.2;q25) qui fait intervenir deux gènes : le gène TFE3 sur le chromosome X (Xp11.2) et le gène APSCR1 sur le chromosome 17 (17q25) aboutissant à la formation d’un facteur chimérique de transcription [7]. Des études récentes suggèrent que les analyses en FISH avec sonde TFE3 seraient plus sensibles et plus spécifiques que l'immunohistochimie, et pourraient donc être considérées comme le gold standard pour la recherche de ce transcrit [4]. Une autre translocation, YAP1-TFE3, a été récemment rapportée [4]. Le grade histo-pronostique selon la Fédération nationale des centres de lutte contre le cancer (FNLCC) ou le Children Oncology Group (COG) semble peu représentatif du potentiel évolutif de la tumeur [4]. Les facteurs pronostiques sont représentés par l'âge (meilleur pronostic chez l'enfant), la taille tumorale (plus mauvais pronostic pour les tumeurs supérieures à 5 cm) et la présence de métastases au moment du diagnostic [3]. Le SAPM présente une évolution souvent indolente, avec une survie à long terme malgré un taux élevé de métastases pulmonaires et cérébrales [7]. Les récidives sont rares après exerese complète, mais les métastases sont fréquentes. Elles peuvent être tardives après une longue période de rémission ou précoces au moment du diagnostic ou parfois même être révélatrices du sarcome [3].
Le sarcome alvéolaire des parties molles est une tumeur rare qui touche avec prédilection le sujet jeune et qui peut être révélé par des métastases. Il est caractérisé par une anomalie moléculaire spécifique. L’aspect morphologique est généralement évocateur basé sur l’aspect alvéolaire et confirmé par l’étude immuno-histochimique à l’aide d’anticorps anti TFE3. Le recours à la biologie moléculaire est parfois nécessaire pour les cas difficiles.
- Portera C, Ho V, Patel S, Hunt K, Feig B, Respondek P, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91:585-91 pubmed
- Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. In: World Health Organization Classification of Tumours of Soft Tissue and Bone. Lyon: IARC 2013.