Introduction : Le rhabdomyosarcome est une tumeur maligne extrêmement agressive dont la localisation orbitaire est très rare. Nous rapportons le cas clinique d’un rhabdomyosarcome orbitaire chez l’enfant. Observation : Un enfant âgé de 13 ans avait présenté une exophtalmie droite isolée d’évolution progressive. L’examen clinique avait objectivé une exophtalmie droite isolée et le reste de l’examen clinique était sans particularité. La tomodensitométrie a mis en évidence un processus tumoral intra-orbitaire droit. L’analyse histologique a révélé un rhabdomyosarcome embryonnaire. Le traitement associant une chimiothérapie à base d’ifosfamide, de vincristine et d’actinomycine, et une radiothérapie orbitaire de 50 Gy selon la technique de modulation d’intensité (IMRT), a permis d’obtenir une rémission complète avec un recul de 6 mois. Conclusion : Le rhabdomyosarcome est une tumeur rare. La précocité de la prise en charge améliore le pronostic vital et fonctionnel.
Introduction: Rhabdomyosarcoma is a very aggressive tumor which is rarely localized in the orbit. We report a case of an orbital rhabdomyosarcoma in a child. Case: A 13-years-old male patient presented a right exophthalmia. Physical examination demonstrated an isolated right exophthalmia and the rest of the clinical examination was normal. CT showed a right intraorbital tumor process. Pathological examination revealed the diagnosis of embryonic rhabdomyosarcoma. Treatment by chemotherapy combining ifosfamide, vincristine and actinomycin, and orbital radiotherapy (50 Gy) allowed a complete remission with a 6 months’ follow-up. Conclusion: Rhabdomyosarcoma is a rare tumor. Early management improves vital and functional prognosis.
Le rhabdomyosarcome est une tumeur rare, qui représente 5 % des tumeurs survenant chez l’enfant [1]. Sa localisation est orbitaire dans 10 % des cas [1] et la localisation palpébrale est exceptionnelle [2]. Il s’agit d’une tumeur maligne extrêmement virulente dont le diagnostic précoce améliore considérablement la survie et le pronostic visuel [3]. Nous rapportons le cas d’un rhabdomyosarcome orbitaire droit présenté sous la forme d’une exophtalmie droite isolée d’évolution chez un enfant âgé de 13 ans.
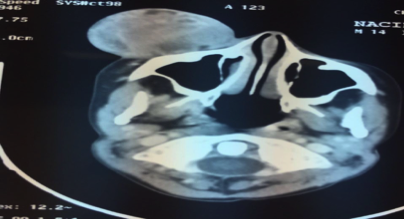
Nous rapportons l’observation d’un enfant âgé de 13 ans, sans antécédents pathologiques notables, qui présente depuis un mois une exophtalmie droite d’évolution progressive. Le reste de l’examen somatique est sans particularité. Une TDM cervico faciale en faveur d’un processus d’allure tumorale intra conique sus et rétro -oculaire droit hyperdense et hétérogène après injection de produit de contraste, mesurant 31 x 29 mm en hauteur, ce processus est en contact intime avec la postéro-interne du globe oculaire, sans liseré graisseux de séparation et sans bourgeon endo-oculaire visible ,il est responsable d'un refoulement du globe oculaire avec exophtalmie grade 2 (Figure 1).
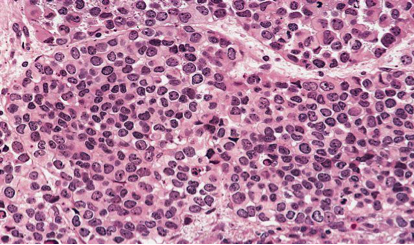
Il englobe et étire le muscle droit supérieur et les autres muscles droits ainsi que le nerf optique sont respectés. Il n’était pas visualisé de lésion osseuse lytique ou de prise de contraste suspecte aux niveaux cérébral et méningé un bilan d’extension fait d’un scanner thoraco abdomino pelvien était normal. Une biopsie a été faite et l’examen histologique et immunohistochimique était en faveur d’un rhabdomyosarcome alvéolaire type C (Figure 2).

Compte tenu de la taille de la tumeur et du risque fonctionnel local, la chirurgie été récusée une chimiothérapie fut débutée selon le protocole RMS 2005, qui consiste en 5 cures associant de l’ifosfamide, de la vincristine et de l’actinomycine (IVA). Un bilan de réponse au traitement fut réalisé au bout de trois cures : le scanner orbitaire mit une évidence une nette diminution en taille du processus tumoral inta et extra orbitaire droit, centré sur la paroi supérieure de l'orbite et développé au dépend du muscle du muscle droit supérieur, de densité tissulaire, rehaussé après contraste et mesurant 29*23*13 mm versus 54*55*54 mm Devant la persistance de ce résidu tumorale : 3cures d’IVA sont rajoutés, une IRM orbitaire d’évaluation a été faite après la 8ème cure d’IVA était en faveur d’un très petit résidu centrée sur le muscle droit supérieur de l'orbite droit sans syndrome de masse ou de prise de contraste pathologique vue uniquement sur deux coupes de la TDM de contrôle(Figure 3).
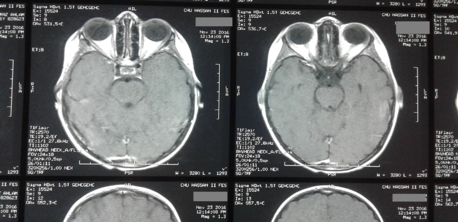
Une radiothérapie externe par modulation d’intensité (IMRT) a été indiquée après la chimiothérapie, sur le résidu tumoral, chez ce patient à la dose totale de 50 Gy : un fractionnement classique 2Gy/fr en 25 séances, avec une bonne tolérance clinique à la radiothérapie externe. Une IRM orbitaire faite 6 mois après la radiothérapie externe, était en faveur d’un petit épaississement inflammatoire post radique du muscle droit supérieur, sans prise de contraste pathologique (Figure 4).
Le rhabdomyosarcome est une tumeur rare (5 % des tumeurs de l’enfant) dont la localisation orbitaire est la plus fréquente (10 % des cas) [1]. Cette tumeur imitant du tissu musculaire strié et se développant à partir des cellules embryonnaires mésenchymateuses peut survenir aussi bien aux dépens d’un muscle strié que dans une zone en étant dépourvue [3]. Ses localisations préférentielles sont par ordre de fréquence la tête et le cou (40 %), l’appareil génito-urinaire (20 %), les extrémités (20 %) et le tronc (10 %) [3]. parmi les localisations orbitaires, 76 % se situent dans la cavité orbitaire ; 12 % sont conjonctivales, 9 % intraoculaires, et seulement 3 % sont palpébrales [2], ce qui distingue ce cas des présentations habituelles. Les localisations intraoculaires sont exceptionnelles et se développent à partir du corps ciliaire ou de l’iris [4-9]. D’un point de vue radiologique, l’imagerie n’est pas pathognomonique, mais fournit des arguments en faveur du diagnostic [10] : densité tissulaire de la lésion, réhaussement après injection d’iode ou de gadolinium, aspects d’ostéolyse des parois orbitaires. Elle permet de visualiser la lésion, de la localiser dans l’orbite, parfois d’en affirmer la structure d’origine, de la mesurer, d’identifier ses rapports et d’en préciser l’extension orbitaire ou encéphalique. Cette observation illustre, par la normalité de l’imagerie initiale, la croissance tumorale très rapide [11, 12] du rhabdomyosarcome qui en fait une urgence. Les différentes formes histologiques du rhabdomyosarcome sont embryonnaires, alvéolaires, pléomorphiques qui sont rarement localisées à l’orbite [2]. L’apport de l’immunohistochimie est précieux dans ce type de tumeur et permet de redresser certains diagnostics erronés. En effet, la distinction entre le rhabdomyosarcome et d’autres tumeurs mésenchymateuses est parfois difficile surtout lorsqu’il s’agit de formes indifférenciées, comme dans cette observation. La prise en charge qui peut comporter une chimiothérapie, une chirurgie, et/ou une radiothérapie, nécessite une concertation multidisciplinaire. La chirurgie n’est pas systématique. L’Intergroup Rhabdomyosarcoma Study Group I (IRSG I) fait prévaloir la préservation de la fonction par rapport à une résection complète [13]. Ainsi, en raison du volume tumoral initial et afin d’éviter une exérèse mutilante, il a été décidé de ne pas avoir recours à la chirurgie chez cet enfant. Le rhabdomyosarcome est une tumeur chimio-sensible. Les protocoles actuels combinent trois molécules lorsque la tumeur n’est pas métastatique : la vincristine, l’actinomycine et le cyclophosphamide ; la vincristine, l’actinomycine et l’ifosfamide ; la vincristine, l’étoposide et l’ifosfamide. Une efficacité supérieure de la combinaison vincristine et actinomycine a été démontrée [14]. Le rhabdomyosarcome est également une tumeur radiosensible, mais qui nécessite des doses élevées non dénuées d’effets indésirables. Les principaux effets indésirables sont la cataracte radique (55 %), la sécheresse oculaire (36 %), l’hypoplasie orbitaire (24 %), le ptôsis (9 %), et la rétinopathie radique (90 %) [8]. Chez cet enfant, l’irradiation n’a été entreprise que lorsque l’imagerie a confirmé l’existence d’un résidu tumoral après 6 séances de chimiothérapie. En effet, cette technique n’altère pas la survie des patients et en préserve un certain nombre des effets indésirables de la radiothérapie [15].
Depuis l’introduction du traitement multimodal par chimiothérapie, chirurgie et/ou radiothérapie, le pourcentage de survie des patients atteints de rhabdomyosarcomes s’est nettement amélioré. Sur le plan local, une régression est constatée dans 80 % des cas (20 % de récurrences). Sur le plan régional, aucune diffusion ganglionnaire n’est constatée dans 94 % des cas ; 6 % des cas apparaissent des adénopathies. Enfin, sur le plan général, 94 % des patients ne présentent pas de diffusion systémique contre 6 % des cas où apparaissent des métastases viscérales. Au total, la survie à 5 ans est de 94 % pour la forme embryonnaire et de 74 % pour la forme alvéolaire [11]. La survie dépend en particulier du caractère non métastatique de la maladie. À cela s’ajoute le risque local fonctionnel, en particulier d’amblyopie et de compression du nerf optique : le rhabdomyosarcome est donc une urgence diagnostique et thérapeutique [12-16], d’autant plus qu’il s’agit d’une tumeur de développement brutal [11, 12]. Les diagnostics différentiels sont nombreux et sont regroupés en deux catégories : les étiologies tumorales et non-tumorales [10-17]. Les étiologies tumorales comprennent les tumeurs kystiques (kystes dermoïdes, carcinomes embryonnaires), les tumeurs vasculaires (hémangiome capillaire ou hémangioendothéliome bénin du nourrisson, lymphangiomes), les tumeurs nerveuses (neurofibromes orbitaires, neurofibrome plexiforme, gliome du nerf optique et du chiasma), les tumeurs de l’os et du cartilage (dysplasie fibreuse, kystes anévrismaux, fibromes ossifiants juvéniles, ostéosarcomes, chondrosarcomes), les maladies histiocytaires (histiocytose à cellules de Langerhans ou histiocytose X, xanthogranulome juvénile), les atteintes orbitaires aux cours des hémopathies (lymphomes dont le lymphome de Burkitt, localisation tumorale au cours d’une leucémie), les métastases (neuroblastome, sarcome d’Ewing), et enfin, les tumeurs propagées à l’orbite dont le rétinoblastome qui occupe une place encore importante dans les pays où l’accès aux soins est plus difficile [17]. Les étiologies non-tumorales sont les mucocèles, les méningocèles, les encéphalocèles et microphtalmies avec kystes, les pseudo-tumeurs inflammatoires, et les infections (dacryocystite, ethmoïdite, cellulite orbitaire, abcès orbitaire).
Cette observation est atypique en raison de la localisation orbitaire de la tumeur, mais également le rhabdomyosarcome est un diagnostic auquel il faut songer afin d’instaurer une prise en charge précoce pour un meilleur pronostic vital et fonctionnel.
- Le Gall F, Edan C, Toulemont P, Jouan H, Urvoy M, Ramee M. [Orbital rhabdomyosarcoma in children. Apropos of 2 cases]. J Fr Ophtalmol. 1994;17:67-73 pubmed
- Shields C, Shields J, Honavar S, Demirci H. Clinical spectrum of primary ophthalmic rhabdomyosarcoma. Ophthalmology. 2001;108:2284-92 pubmed
- Maurer H, Beltangady M, Gehan E, Crist W, Hammond D, Hays D, et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer. 1988;61:209-20 pubmed
- Wilson M, McClatchey S, Zimmerman L. Rhabdomyosarcoma of the ciliary body. Ophthalmology. 1990;97:1484-8 pubmed
- Elsas F, Mroczek E, Kelly D, Specht C. Primary rhabdomyosarcoma of the iris. Arch Ophthalmol. 1991;109:982-4 pubmed
- Woyke S, Chwirot R. Rhabdomyosarcoma of the iris. Report of the first recorded case. Br J Ophthalmol. 1972;56:60-4 pubmed
- Font R, Zimmerman L. Electron microscopic verification of primary rhabdomyosarcoma of the iris. Am J Ophthalmol. 1972;74:110-7 pubmed
- Shields J, Shields C. Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol. 2003;48:39-57 pubmed
- Jones I, Reese A, Kraut J. Orbital rhabdomyosarcoma. An analysis of 62 cases. Am J Ophthalmol. 1966;61:721-36 pubmed
- Kodet R, Newton W, Hamoudi A, Asmar L, Wharam M, Maurer H. Orbital rhabdomyosarcomas and related tumors in childhood: relationship of morphology to prognosis--an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol. 1997;29:51-60 pubmed
- Ducrey N, Spahn B. [Emergencies in nontraumatic orbital diseases]. J Fr Ophtalmol. 2002;25:927-30 pubmed
- Crist W, Anderson J, Meza J, Fryer C, Raney R, Ruymann F, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19:3091-102 pubmed
- Rousseau P, Flamant F, Quintana E, Voute P, Gentet J. Primary chemotherapy in rhabdomyosarcomas and other malignant mesenchymal tumors of the orbit: results of the International Society of Pediatric Oncology MMT 84 Study. J Clin Oncol. 1994;12:516-21 pubmed
- Ducrey N, Nenadov-Beck M, Spahn B. [Update of orbital rhabdomyosarcoma therapy in children]. J Fr Ophtalmol. 2002;25:298-302 pubmed
- Poso M, Mwanza J, Kayembe D. [Malignant tumors of the eye and adnexa in Congo-Kinshasa]. J Fr Ophtalmol. 2000;23:327-32 pubmed
- Belmekki M, El Bakkali M, Abdellah H, Benchrifa F, Berraho A. [Epidemiology of orbital processes in children. 54 cases]. J Fr Ophtalmol. 1999;22:394-8 pubmed