Introduction : Les mucopolysaccharidoses (MPS) constituent un groupe hétérogène de maladies métaboliques héréditaires, liées à un déficit enzymatique lysosomial responsable d’une maladie de surcharge par accumulation de glycosaminoglycanes (GAG), anciennement appelées mucopolyaccharides. Les auteurs rapportent les cas de 27 enfants, colligés au sein de l’unité de neurologie pédiatrique de Rabat. Objectif : Le but de ce travail est de dégager les données cliniques, biologiques et radiologiques de cette affection caractérisée par une très grande hétérogénéité clinique et biologique. Résultats : Notre série comporte quinze garçons et douze filles avec un sex-ratio de 1,25. L’âge moyen était de 7 ans et demi. La consanguinité des parents a été retrouvée dans 52% des cas. L’antécédent de cas similaire dans la famille a été retrouvé dans 44% des cas. Les circonstances de découverte de la maladie étaient : une déformation rachidienne dans 50% des cas, un retard psychomoteur dans 29% des cas, une distension abdominale dans 4% des cas, une hernie ombilicale dans 11% des cas et une hernie inguinale dans 6% des cas. La tomodensitométrie cérébrale a objectivé un aspect hypodense de la substance blanche sus-tentorielle associée à une ventriculomégalie dans neuf cas. Elle était sans particularité dans dix-huit cas. Le bilan radiographique du crâne, du squelette et des membres, a montré des anomalies radiologiques dans 63% des cas. Le dosage urinaire des GAGS a objectivé des valeurs augmentées dans tous les cas. La prise en charge des enfants a consisté en une rééducation fonctionnelle et motrice, et un traitement cardio-sélectif en cas d’atteinte cardiaque. Un traitement enzymatique substitutif à base d’aldurazyme* a été administré dans un seul cas de MPS de type 1. L’évolution était marquée par le décès d’un cas dans un tableau infectieux, et l’absence d’aggravation clinique dans les autres cas, avec un recul moyen de trois ans et demi. Conclusion : Les mucopolysaccharidoses constituent un groupe hétérogène de maladies métaboliques héréditaires relativement rares. Leur traitement reste symptomatique vu le coût et la non disponibilité du traitement enzymatique substitutif au Maroc. Ainsi, la seule alternative qui reste pour diminuer l’incidence de ces affections est le diagnostic prénatal réalisé au sein des familles à risque.
Introduction: Mucopolysaccharidoses (MPS) are a heterogeneous group of inherited metabolic diseases related to a lysosomal enzyme deficiency which causes the accumulation of glycosaminoglycans (GAGs). The authors present 27 cases collected in the pediatric neurology unit of Rabat. Objective: The aim of this work was to identify the clinical, biological and radiological features of this disorder. Results: Our series included fifteen boys and twelve girls with a sex ratio of 1.25. The mean age was 7 years and a half. Parental consanguinity was found in 52% of cases. The history of similar cases in the family was found in 44% of cases. The circumstances of disease discovery were: spinal deformity in 50% of cases, psychomotor retardation in 29% of cases, abdominal distension in 4% of cases, umbilical hernia in 11% of cases and inguinal hernia in 6% of cases. CT brain showed a hypodense appearance of the supratentorial white matter associated to ventriculomegaly in nine cases. It was normal in eighteen cases. The skull, skeleton and limbs X-rays showed radiological abnormalities in 63% of cases. Urinary assay GAGS objectified increased values in all cases. The management was based on functional and motor rehabilitation, cardioselective treatment in the case of cardiac involvement. Enzyme replacement therapy based on Aldurazyme* was given only in one case of type 1 MPS. The evolution was marked by a death in one case by infection, and the absence of clinical worsening in the remaining cases with a mean follow-up of 3.5 years. Conclusion: Mucopolysaccharidoses are a heterogeneous group of relatively rare hereditary metabolic diseases. The treatment options are limited because of the cost and unavailability of enzyme replacement therapy in Morocco. Thus, prenatal diagnosis in high-risk families remains the only way to decrease the incidence of these disorders.
Les mucopolysaccharidoses (MPS) sont des maladies de surcharge lysosomale. Elles sont dues à un déficit enzymatique, aboutissant à une accumulation intralysosomale de glycosaminoglycanes (GAG) anciennement appelés mucopolysaccharides acides. Cette surcharge entraîne une excrétion accrue des GAG dans les urines [1]. Une grande hétérogénéité clinique et biologique caractérise les mucopolysaccharidoses, ce qui rend leur diagnostic difficile. Leur transmission est autosomique récessive, à l'exception de la maladie de Hunter (MPS II) transmise sur le mode récessif lié au chromosome X. On en distingue sept types cliniques qui correspondent à 11 déficits enzymatiques différents. Chaque maladie est caractérisée par une grande variabilité concernant l’âge de début et le degré d’atteinte des différents organes [2]. A l’exception de la maladie de Morquio, dont le tableau clinique est distinct, les MPS sont caractérisées par une dysmorphie et des lésions osseuses similaires bien que la sévérité est différente. Le traitement de ces maladies handicapantes est essentiellement symptomatique, il est onéreux car il nécessite l’intervention de différentes unités de plusieurs spécialistes : psychologues, pédiatres, chirurgiens orthopédistes, orthophonistes, kinésithérapeutes, cardiologues, odontologistes et biologistes [3].
- • Période : étude rétrospective étalée sur 12 ans allant de janvier 2002 à janvier 2014.
- • Lieu : Unité de neurologie pédiatrique-maladies métaboliques. Service de pédiatrie II, hôpital d’enfant de Rabat.
- • Patients : tous les malades suspects cliniquement ou radiologiquement d’être atteints d’une mucopolysaccharidose, et ayant bénéficié d’un bilan biologique de certitude confirmant ce diagnostic.
- • Les patients n’ayant pas bénéficié d’un bilan de certitude (ou en cours de diagnostic).
- • Les patients dont le diagnostic de mucopolysaccharidose a été établi par d’autres services.
Nous avons réussi à collecter les données cliniques, paracliniques , thérapeutiques et évolutives, concernant les malades répondant aux critères d’inclusion et nous avons regroupé les différents paramètres sous forme de fiches de renseignements individuelles. La collecte de données a été établie :
- *en consultant les dossiers médicaux disponibles aux archives du service de pédiatrie II de l’hôpital d’enfants de Rabat.
- *en contactant les familles des malades dont on dispose d’un numéro de téléphone ou d’une adresse.
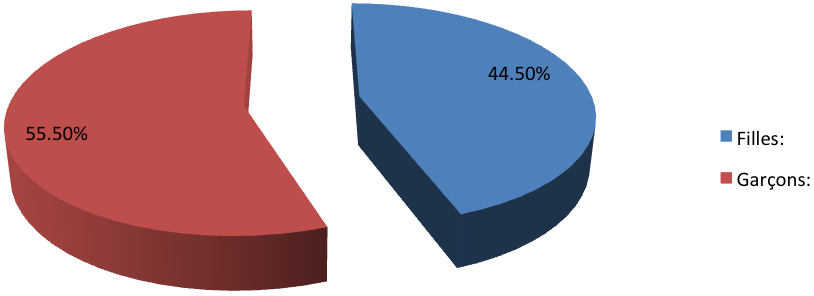
Notre série comporte quinze garçons et douze filles avec un sex-ratio de 1,25. L’âge moyen était de 7 ans et demi. La consanguinité des parents a été retrouvée dans 52% des cas. L’antécédent de cas similaire dans la famille a été retrouvé dans 44% des cas.
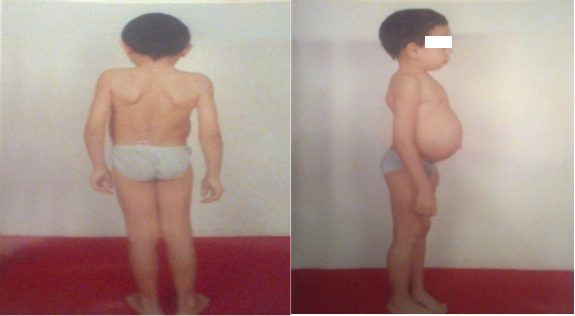
Les circonstances de découverte de la maladie étaient : Une déformation rachidienne dans 50% des cas, un retard psychomoteur dans 29% des cas, une distension abdominale dans 4% des cas, une hernie ombilicale dans 11% des cas et une hernie inguinale dans 6% des cas. L’examen clinique trouvait un retard statural dans 48% des cas, et une dysmorphie faciale dans 70% des cas. La macrocranie et la macroglossie étaient présentes dans 15% des cas. Les anomalies dentaires existaient dans 30% des cas, et les anomalies thoraco-rachidiennes dans 41% des cas.

L’hyperlaxité ligamentaire a été présente dans 4% des cas, et la raideur articulaire dans 18,5% des cas. Les anomalies cardio-respiratoires ont été retrouvées dans 55% des cas. Les anomalies viscérales ont été représentées par une hernie ombilicale dans 26% des cas, une hernie inguinale dans 18,5% des cas, une splénomégalie dans 18,5% des cas, et une hépatomégalie dans 22% des cas. Les anomalies ophtalmiques ont été présentes dans 33% des cas, et les végétations adénoïdes dans 22% des cas. La TDM cérébrale a objectivé un aspect hypodense de la Substance Blanche sus-tentorielle associée à une ventriculomégalie dans neuf cas, et était normale dans dix-huit cas. (Figure 3)

Le bilan radiographique du crane, du squelette et des membres, a montré des anomalies radiologiques dans 63% des cas :
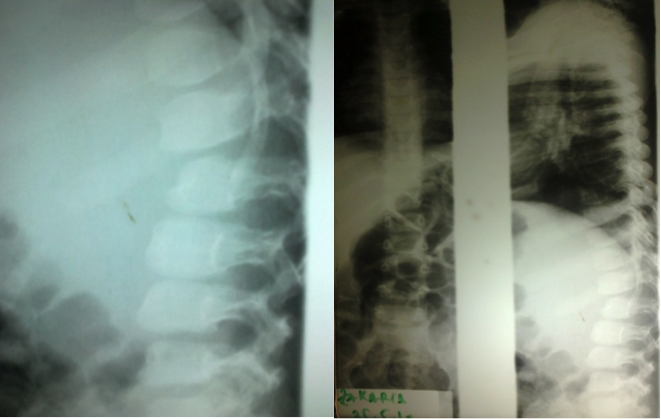
- *La radiographie du crane, a montré une scaphocéphalie avec un aspect en « Oméga » de la selle turcique (Figure 4) dans huit cas, une craniosténose dans un cas, était normale dans dix cas, et n’a pas été faite dans sept cas.
- *La radiographie thoracique a montré un aspect affilé des côtes dans huit cas, un aspect élargi des cotes en palette dans un cas, était normale dans douze cas, et n’a pas été faite dans six cas.
- *La radiographie du rachis a objectivé une scoliose dans cinq cas, une cyphose dans un cas, une hypoplasie des coins antéro-supérieurs de L2 (Figure 5) dans quatre cas, un prolongement des corps vertébraux en longuettes dans deux cas, une platispondylie dans un cas, était normale dans dix cas, et n’a pas été faite dans quatre cas.
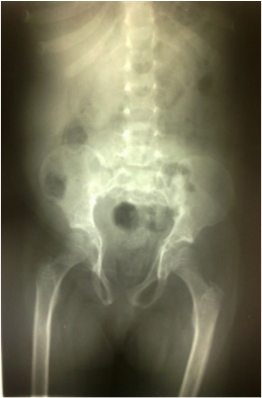
- *La radiographie du Bassin a montré un bassin étroit avec déformation des cols fémoraux dans deux cas, une dysplasie cotyloïdienne bilatérale avec coxa-plana dans deux cas, une coxa-valga dans trois cas, un aspect grêle des branches ischio et ilio-pubiennes (Figure 6) dans deux cas, une horizontalisation des toits de cotyles dans un cas, était normale dans quinze cas, et n’a pas été faite dans un cas.
- *La radiographie des membres a montré un aspect conique de la phalange du pouce dans un cas, un aspect court des métacarpiens dans un cas, et était normale dans le reste des cas.
Le dosage urinaire des GAGS a objectivé des valeurs augmentées dans tous les cas, et l’électrophorèse des MPS a montré la présence de bandes anormales, en faveur de la maladie d’HURLER dans 44% des cas, de la maladie de de morquio dans 44% des cas, et de la maladie de sanphilippo dans 12% des cas. Le dosage enzymatiques de l’Alpha Iduronidase était effondré (=0 nM/h/mg) dans douze cas, confirmant le diagnostic de la maladie d’HURLER.
L’étude génétique faite dans un seul cas, a objectivé la présence de la mutation délétère du gène IDUA (c.3233C>G ; p.Pro533Arg) à l’état homozygote. La prise en charge des enfants a consisté en une rééducation fonctionnelle et motrice, un traitement cardiosélectif en cas d’atteinte cardiaque. Un traitement enzymatique substitutif à base d’aldurazyme* a été utilisé dans un seul cas de MPS type 1. L’évolution avec un recul de trois ans et demi, était marquée par le décès d’un cas dans un tableau infectieux, et l’absence de l’aggravation clinique dans le reste des cas.

Les mucopolysaccharidoses constituent un groupe hétérogène de maladies métaboliques héréditaires relativement rares.
Un déficit enzymatique lysosomial, génétiquement déterminé, est à l'origine de l'incapacité de ces organites à dégrader, étape par étape, ces macromolécules sulfatées et à libérer dans le cytosol les produits de leur catabolisme [4].
| MPS | Nom | Localisation | Enzyme | ES |
|---|---|---|---|---|
| I | (Hurler/Scheie) | 4p16.3 | a-l-Iduronidase | + |
| II | Hunter | Xq27.3-q28 | Iduronate-2-sulfatase | + |
| IIIA | San Philippo | 17q25.3 | Héparane sulfate sulfatase | |
| IIIB | San Philippo | 17q21.1 | a-N-Ac-d-glucosaminidase | |
| IIIC | San Philippo | Xs 8 | a-glucosaminide-N-Ac-transférase | |
| IIID | San Philippo | 12q14 | N-Ac-glucosamine-6-sulfatase | |
| IVA | Morquio | 16q24.3 | Galactose-6-sulfatase | |
| IVB | Morquio | 3p21.33 | þ-galactosidase | |
| VI | Maroteaux-Lamy | 5p11.13 | arylsulfatase B | + |
| VII | Sly | 7q21q.22 | þ-glucuronidase | |
| IX | 3p21.3p21.2 | Hyaluronidase |
La MPS I est due à un déficit enzymatique en α-L-iduronidase. Ceci est compatible avec notre étude. Une affection attribuée au même déficit enzymatique, mais avec une traduction clinique très différente, est aujourd’hui connue sous le nom de maladie de Scheie (MPS IS) [6]. La mucopolysaccharidose de type II ou syndrome de Hunter, est due à un déficit enzymatique lysosomal en iduronate-2-sulfatase, entrainant une accumulation progressive de glycosaminoglycanes (héparane sulfate et dermatane sulfate) dans tous les tissus de l’organisme [7]. La mucopolysaccharidose de type VI (MPS VI) ou maladie de Maroteaux-Lamy est due à un déficit en N-acétylgalactosamine- 4-sulfatase ou arylsulfatase B, normalement responsable du catabolisme du dermatane sulfate [7]. Pour la MPS de type IV, le déficit de quatre enzymes nécessaires à la dégradation de l’héparane sulfate est responsable des différents sous-types de MPS III : heparan N-sulfatase pour la MPS IIIA, alpha-N- acétylglucosaminidase pour la MPS IIIB, acetyl-CoA: a-glucosaminide acetyltransferase pour la MPS IIIC, et N-acétylglucosamine-6-sulfatase pour la MPS IIID [8]. La transmission des mucopolysaccharidoses est autosomique récessive, à l'exception de la maladie de Hunter (MPS II) transmise sur le mode récessif lié au chromosome X. Cette maladie n’a pas été représentée dans notre travail. Les MPS sont suspectées cliniquement devant un intervalle libre, une dysmorphie, une hypertrichose, une cyphose lombaire, des doigts en griffe, flessum, une hépatosplénomégalie, et une anomalie de croissance. Ceci est compatible avec notre étude. Leurs manifestations cliniques sont résumées dans le tableau 2.
| Maladie | Retard mental | Dysmorphie | Hépato splénomégalie | Opacités cornéennes | Perte d’audition | Dysostose multiple |
|---|---|---|---|---|---|---|
| MPS IH | + | ++ | + | + | + | +++ |
| MPS IS | - | + | + | ± | ||
| MPS IH/S | ± | + | + | + | ++ | |
| MPS II sévère | + | + | + | - | ++ | |
| MPS II modérée | - | - | + | |||
| MPS IIIA | + | + | + | ± | ± | |
| MPS IV A | - | ± | + | ± | Dysplasie spondyloépiphysaire | |
| MPS VI | - | ± | + | + | + | +++ |
| MPS VII | + | + | + | + à +++ |
- * Les maladies de Hürler-Scheie, Hunter, Sly, se caractérisent par une atteinte multisystémique évolutive :
- Ophtalmologique avec mégacornée, dépots cornéens, glaucome, rétinopathie, oedème papillaire, et atrophie optique.
- Cardiaque avec valvulopathie (osler++), cardiomyopathie, coronaropathie, hypertension artérielle, et hypertension artérielle pulmonaire.
- Respiratoire avec encombrement chronique et surinfections, infiltration pharyngo laryngo-trachéo-bronchique, pneumopathie interstitielle, et un syndrome respiratoire obstructif et restrictif.
- Stomatologique avec retard de poussée dentaire, dents pointues, émail anormal, hypertrophie gingivale, et des kystes.
- Abdomino-digestive avec hernies inguinales, ombilicale, hépato-splénomégalie, diarrhée, constipation, et troubles de déglutition.
- ORL avec une otite séro-muqueuse et une surdité.
- Ostéo-articulaire avec cyphose thoraco-lombaire, genu valgum, dysostoses multiples, enraidissement articulaire, cassure staturopondérale puis petite taille.
- Neurologique avec retard /régression psychomotrice, agitation, syndrome du canal carpien, compression médullaire, hydrocéphalie, atrophie cérébrale, épilepsie, tétraparésie spastique, et troubles du sommeil [9].
- * La mucopolysaccharidose de type VI (MPS VI) ou maladie de Maroteaux-Lamy est caractérisée cliniquement par des lésions squelettiques avec petite taille, dysostosis multiplex et enraidissement articulaire, une atteinte valvulaire cardiaque et respiratoire, une hépatosplénomégalie, des sinusites et des otites moyennes, des apnées du sommeil, des opacités cornéennes, un syndrome du canal carpien et des hernies inguinales et ombilicales. Les patients peuvent aussi présenter une hydrocéphalie, une atrophie optique avec cécité et un risque de compression de leur moelle cervicale par instabilité ou épaississement méningé au niveau de la colonne vertébrale cervicale [7].
- * Pour la mucopolysaccharidose de type III (MPS III) ou maladie de Sanfilippo, les premiers symptômes de la maladie, apparaissent entre 3 et 6 ans: une agressivité, un trouble de comportement contrastant avec une atteinte somatique modérée, et un retard de développement portant sur le langage. La régression intellectuelle est rapide et sévère. L'atteinte somatique est au contraire modérée avec au début une macrocéphalie et une avance staturopondérale; la dysmorphie faciale est modérée ou absente ; les cheveux sont parfois épais et drus ; l'atteinte squelettique est discrète. Une hépatosplénomégalie est rare. Une surdité profonde est classique même dans les formes modérées. A partir de 10 ans, les enfants sont généralement plus calmes, développent des troubles orthopédiques, des troubles de l'alimentation et du transit, des troubles du sommeil (insomnies), des convulsions, et les surinfections respiratoires sont fréquentes. Dans la phase finale, les malades sont grabataires, perdent tout contact avec leur entourage et développent une démence profonde. Le décès survient habituellement vers 20 ans, souvent au décours d'une infection respiratoire, mais des cas avec survie prolongée ont été rapportés [10].
- * Pour les MPS de type IV ou syndrome de Morquio A, l’apparition de la symptomatologie clinique débute généralement vers l’âge de 2 à 3 ans, et peut se présenter sous différentes formes cliniques en fonction du degré de sévérité. La forme classique sévère est le prototype de la dysplasie spondylo-épiphyso-métaphysaire. On observe un genu valgum, un retard de croissance avec tronc et cou courts et une démarche dandinante [11]. L’atteinte osseuse inclut une platyspondylie, une hypoplasie de l’apophyse odontoïde, une cyphoscoliose et des déformations thoraciques ; la taille définitive n’excède pas 1 mètre. La face est caractérisée par un prognathisme, une grande bouche avec de petites dents espacées et souvent abîmées. Les complications neurologiques sont secondaires aux anomalies squelettiques et peuvent entraîner précocement une myopathie cervicale et des risques de paralysie respiratoire avec mort subite. Les atteintes valvulaires cardiaques sont responsables de la réduction de la durée de vie à une trentaine, voire une vingtaine d’année, dans les formes sévères [12].
À côté de cette forme classique, des formes modérées sont observées, où les patients ont une espérance de vie prolongée, les manifestations cliniques extra-squelettiques étant beaucoup plus tardives. Entre les formes sévères et modérées, on décrit une forme intermédiaire qui se caractérise par une meilleure qualité de vie que celle associée à la forme sévère. Au cours de la MPS IVA, l’intelligence est conservée, ce qui différencie cette forme des autres MPS [12]. Les MPS ont des signes radiologiques communs: Une déformation cunéiforme d’une vertèbre de L1 ou L2, un aspect trapu des os, petites épiphyses, une coxa valga, un cotyle fuyant, un défaut de modelage des métacarpiens et des phalanges, ainsi qu’une scaphocéphalie, et une selle turcique allongée. Ceci est compatible avec notre étude. Le diagnostic biologique est basé sur un prélévement sanguin montrant la présence d’un type particulier de globules blancs (les lymphocytes vacuolés), et sur un prélévement urinaire, montrant un taux élevé de glycoaminoglycanes. Dans les MPS type I, II et III, on trouve un taux élevé de dermatane et d’héparane sulfate, dans les MPS IV, on trouve un taux élevé de kératane sulfate, dans les MPS VI, on trouve un taux élevé de dermatane sulfate, et dans les MPS VII, on trouve un taux élevé de Chondroïtine-S A et C [9]. Le dosage de l’activité enzymatique de l’alpha-L-Iduronidase effondré, apporte la preuve irrévocable du diagnostic de la maladie d’Hurler. Ceci est compatible avec notre étude.
Pour les mucopolysaccharidoses de type I, II et VI, un traitement enzymatique substitutif est disponible. Dans ces trois pathologies, le traitement enzymatique substitutif permet une réduction de l’excrétion urinaire des glycosaminoglycanes urinaires et une amélioration de l’ensemble des tests fonctionnels, test de marche, exploration fonctionnelle respiratoire, et amplitudes articulaires [13]. L’enzymothérapie substitutive est l’unique option thérapeutique actuellement disponible pour traiter les patients atteints de MPS II. Il s’agit d’une enzyme recombinante produite par technologie d’activation de gène à partir d’une lignée de cellules fibroblastiques humaines (idursulfase, Elaprase®) [7]. La transplantation de cellules souches hématopoiétiques (greffe de moelle osseuse) a donné des résultats intéressants dans le traitement des MPS de type VI [14]. Ses indications sont notamment l’existence d’une cardiomyopathie ou d’apnées du sommeil sévères. La greffe prolonge la survie avec amélioration de la qualité de vie, de l’atteinte cardiaque, de la viscéromégalie et de la dysmorphie faciale. En revanche, elle ne parvient pas à corriger ou stabiliser l’atteinte osseuse [7]. Dans notre travail, la prise en charge des enfants a consisté en une rééducation fonctionnelle et motrice, et un traitement cardiosélectif en cas d’atteinte cardiaque. Un traitement enzymatique substitutif à base d’aldurazyme* a été utilisé dans un seul cas de MPS type 1.
Les MPS font partie des maladies de surcharge lysosomiale. Il existe des phénotypes différents. La plupart débutent chez le nouveau né, le nourrisson ou le grand enfant et se traduisent par un large éventail d’atteintes cliniques notamment neurologique.
Elles sont alors source d’un polyhandicap souvent sévère. Les nouveaux traitements offrent l’espoir de prévenir ou d’arrêter le processus pathologique (greffe de moelle osseuse, enzymothérapie substitutive, thérapie génique). Toutefois, Le traitement actuel reste largement symptomatique et multidisciplinaire.
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In : Scriver CR, Beaudet AL, Sly WS, Valle D, et al, eds. The metabolic and molecular bases of inherited disease. 8e édition. New York: McGraw-Hill, 2001: 3421-52.
- Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144:S27-34 pubmed
- L Chkioua, et al. La mucopolysaccharidose de type I : Stratégie diagnostique en Tunisie. Annales de Biologie Clinique. Volume 65, Numéro 2, 175-9, Mars-Avril 2007, Pratique quotidienne.
- Billette de Villemeur T. Le lysosome et les maladies lysosomales. Atlas de l'association Vaincre les maladies lysosomales, Évry, 1998 : 1-3.
- Gérard Chalès, Guillaume Coiffier, Pascal Guggenbuhl. Manifestations ostéoarticulaires des mucopolysaccharidoses et des glycogénoses. Revue du rhumatisme monographies 78 (2011) 254–261.
- Bach G, Friedman R, Weissmann B, Neufeld E. The defect in the Hurler and Scheie syndromes: deficiency of -L-iduronidase. Proc Natl Acad Sci U S A. 1972;69:2048-51 pubmed
- D.P. Germain et al. Thérapies enzymatiques substitutives des maladies lysosomales. La Revue de médecine interne 31 (2010) S279–S288.e3.
- Neufeld E & Muenzer J. The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease 2001:3421–52.
- N. Belmatoug, B. Héron. Les MucoPolySaccharidoses. Centre de Référence des Maladies Lysosomales.
- R.Froissart. Mucopolysaccharidose de type III. Maire Service de biochimie pédiatrique, Hôpital Debrousse 1999 Mise à jour février 2005.
- Ebara S, Kinoshita T, Yuzawa Y, Takahashi J, Nakamura I, Hirabayashi H, et al. A case of mucopolysaccharidosis IV with lower leg paresis due to thoraco-lumbar kyphoscoliosis. J Clin Neurosci. 2003;10:358-61 pubmed
- H Bouzidi, et al. La mucopolysaccharidose IVA (syndrome de Morquio A) : aspects clinique, biologique et thérapeutique Annales de Biologie Clinique. Volume 65, Numéro 1, 5-11, Janvier-Février 2007, Revue générale.
- A. Fouilhoux, N. Guffon. Actualités des traitements enzymatiques substitutifs dans les mucopolysaccharidoses.LaPresseMédicaleVol 36, N° HS1 - mars 2007 96-99.
- Peters C, Steward C. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003;31:229-39 pubmed