Introduction : La leucodystrphie métachromatique (LDM) est une maladie héréditaire autosomique récessive atteignant le système nerveux central et périphérique. Elle est due à un déficit en Arylsulfatase A. Nous rapportons les cas de 4 enfants où le diagnostic de leucodystrophie métachromatique a été retenu sur le dosage de l’activité enzymatique de l’Arylsulfatase A. Objectif : Mettre le point sur cette affection au pronostic sombre. Résultats : Notre série comporte quatre garçons, avec un âge médian de 2 ans et demi. L'examen clinique trouvait une hypertonie pyramidale dans trois cas, une hypotonie généralisée dans un cas, et un strabisme convergent droit dans un cas. La TDM cérébrale a objectivé des images d’hypodensité de la substance blanche dans tous les cas. L’IRM cérébrale a mis en évidence une démyélinisation diffuse et symétrique apparaissant en hyper signal en T2 au niveau de la SB sans atteinte des fibres en U, dans tous les cas. Le dosage de l’activité de l’aryl sulfatase a montré un déficit de l’aryl sulfatase dans 3 cas, et un pseudodéficit dans un cas. La prise en charge des enfants a consisté en un traitement des crises convulsives avec une rééducation fonctionnelle et motrice. Conclusion : La leucodystrophie métachromatique est une maladie neurodégénérative sans traitement spécifique, avec une thérapie enzymatique substitutive à l'étude.
Metachromatic leucodystrphy (MDL) is a recessive autosomal hereditary disease that affects the central and peripheral nervous systems. It is due to deficiency in arylsulfatase A. We report 4 cases of children with the diagnosis of metachromatic leukodystrophy based on the enzymatic activity of arylsulfatase A. Our series includes four boys, with an average age of 2 years and a half. Clinical examination revealed pyramidal hypertension in three cases, generalized hypotonia in one case, and right esotropia in one case. Brain scans identified hypodensity of the white matter in all cases. Brain MRI revealed diffuse symmetrical demyelination with hyper T2 signal in the white matter without U-fibers in all cases. The assay of aryl sulfatase activity showed deficits in aryl sulfatase in 3 cases, and a pseudodeficiency in one case. The management of these cases consisted of the treatment of seizures with functional and motor rehabilitation. Metachromatic leukodystrophy is a neurodegenerative disease with no specific treatment. The enzyme replacement therapy is still being evaluated.
La leucodystrphie métachromatique (LDM) est une maladie héréditaire autosomique récessive atteignant le système nerveux central et périphérique. Elle est due à un déficit en Arylsulfatase A (ASA), qui catalyse la première étape de la dégradation des sulfatides. Cette déficience est responsable d’une accumulation de sulfatide (glycolipides sulfatés, particulièrement les sulfogalactosylcéramides ou sulfogalactocérébrosides) dans le système nerveux et dans d’autres tissus. Elle représente l'une des plus fréquentes LD. Sa fréquence est estimée en Allemagne et en France à 0.6/100000 naissances et en Turquie à 1.43/100000 naissances [1] [2].
L’évolution se fait habituellement vers le décès causé par des infections le plus souvent pulmonaires.
La leucodystrophie métachromatique est suspectée chez des individus présentant des signes de détériorations neurologiques avec des signes de leucodystrophie à l'imagerie par résonance magnétique. Un dosage de l'activité enzymatique de l'arylsulfatase A dans les leucocytes, inférieure à 10 % oriente vers l'origine de la leucodystrophie. Ce diagnostic sera confirmé par une biopsie d’un nerf sensitif et par la présence de sulfatidurie dans les urines.
Ne disposant pas de traitement à l’heure actuelle, tous les espoirs sont tournés vers les perspectives offertes par les thérapies géniques. Les auteurs rapportent quatre cas de leucodystrophie métachromatique, suspectés sur des critères cliniques et radiologiques, et confirmés par un dosage bas de l’activité enzymatique d’Arylsulfatase A.
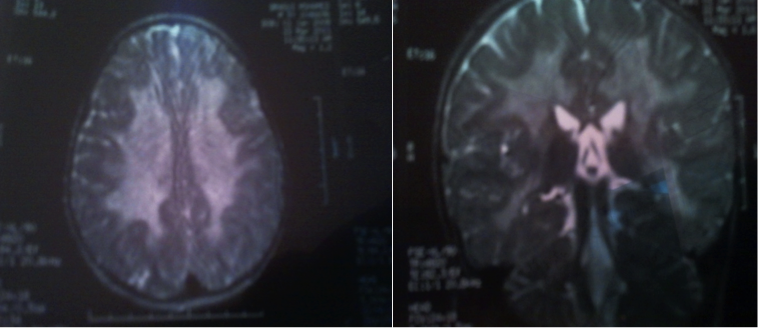
L’enfant M., âgé de 2 ans et demi, est issu de parents non consanguins. L’histoire de sa maladie remontait à 4 mois par l’installation d’une régression psychomotrice avec une dysphagie dans un contexte fébrile. Par ailleurs, l’enfant n’avait pas présenté des crises convulsives. L’examen neurologique trouvait une position assise et une marche non acquises, des membres supérieurs en flexion, une hypertonie pyramidale, une force musculaire générale diminuée, et un Babinski positif. Le poids était à 13kg, la taille à 37cm, et le périmètre crânien à 51cm. La chromatographie des acides aminés dans le sang et dans les urines avait montré une augmentation modérée de l’alanine. L’IRM cérébrale avait mis en évidence la présence d’une anomalie de signal diffuse de la substance blanche en péri ventriculaire (figure 1). La TDM cérébrale avait mis en évidence un aspect hypodense de la substance blanche faisant évoquer un trouble de myélinisation de la substance blanche. L’électroencéphalogramme était normal. L’électromyogramme avait montré un signe de poly neuropathie sensitivomotrice démyélinisante homogène. Le dosage de l’activité enzymatique de l’Arylsulfatase A était effondré (3Nm/H/mg). Ainsi, le diagnostic de leucodystrophie métachromatique est confirmé, et l’enfant a été mis sous piracétam avec rééducation motrice et fonctionnelle.
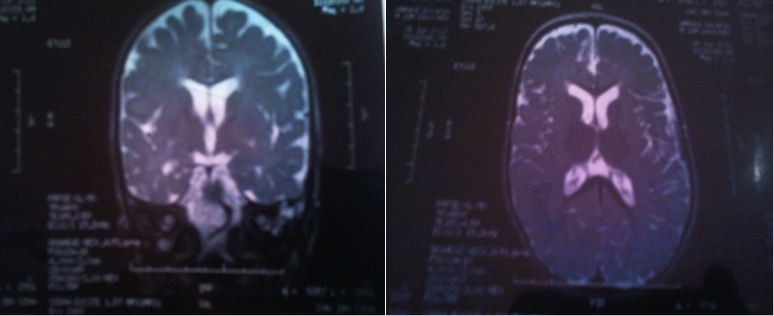
Le nourrisson Z. est âgé de 13 mois, ayant comme antécédents un frère décédé à l’âge de 2 ans ½, issu de parents non consanguins. Le nourrisson a bien évolué jusqu’à l’âge de 4 mois où il est devenu hypotonique, agité, hyper excitable, refusait de téter avec des troubles de déglutition. À partir de l’âge de 4 mois, le nourrisson présentait des crises toniques généralisées, avec déviation des yeux pendant 5 à 10 min avec des spasmes en flexion plusieurs fois/j. L’examen clinique trouvait une hypotonie généralisée, une absence de poursuite du regard, des réflexes ostéo-tendineux présents, un babinski négatif, et des clonies des pieds, avec un strabisme convergent droit. Le périmètre crânien était normal à 47,5cm et la taille était normale à 75cm. L’IRM cérébrale avait objectivé un hypersignal de la substance blanche périventriculaire de façon symétrique, une atteinte des faisceaux en U, avec élargissement des sillons corticaux et des ventricules latéraux (figure 2).
L’électroencéphalogramme a mis en évidence un tracé symétrique bien organisé, pauvre en images physiologiques en rapport avec l’âge avec un rythme de fond normal pour l’âge et une bonne activité postérieure. La SLI ne modifie pas le tracé. Présence de grapho-éléments anormaux faits de foyer temporal droit. L’examen ophtalmologique au fond d’œil a montré des vaisseaux légèrement greliques et tortueux. Le dosage de l’activité enzymatique de l’Arylsulfatase A était diminué à 7 Nm/H/mg, en faveur d’un pseudo-déficit de l’Arylsulfatase A. Le nourrisson a été mis sous rééducation motrice. L’évolution avec un recul de onze mois a été marquée par la survenue d’un décès à l’âge de 2 ans.
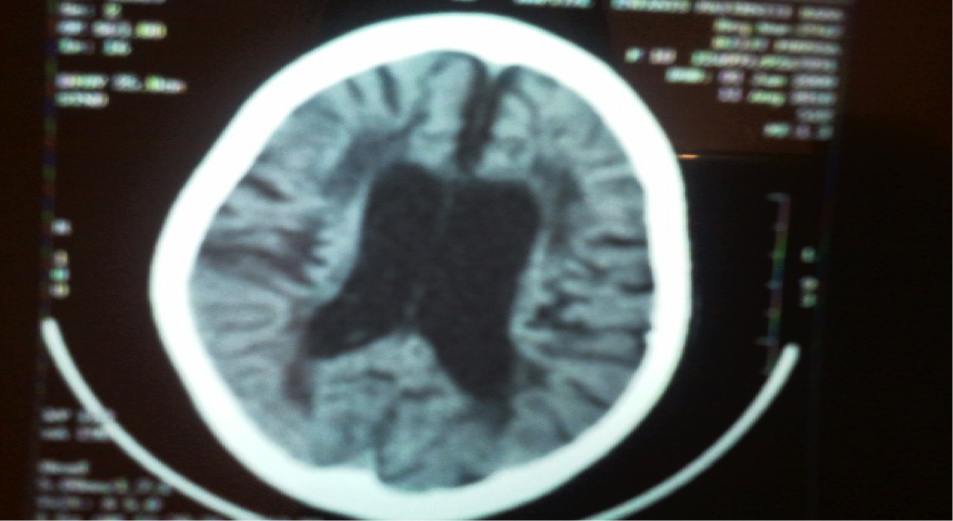
L’enfant M., âgé de 3 ans, est adressé pour une régression psychomotrice. L’enfant était issu de parents consanguins de premier degré, et a une sœur décédée à l’âge de 4 ans dans un tableau d’infirmité motrice cérébrale. L’enfant a présenté à l’âge de 6 ans, sa première crise tonico-clonique généralisée avec révulsion des yeux et mousse aux lèvres dans un contexte d’apyrexie, évoluant par des crises chaque 3 mois. Il a présenté une régression psychomotrice débutant à l’âge de 6 ans suite à un état de mal épileptique sans atteinte intellectuelle, commençant par une monoparésie d’un membre inférieur, puis une perte motrice progressive puis une perte du langage. L’examen clinique trouvait des réflexes vifs avec un babinski négatif. La TDM cérébrale a mis en évidence une hydrocéphalie quadri ventriculaire avec hypodensité de la substance blanche périventriculaire et élargissement des sillons corticaux et vermiens (figure 3).
L’IRM cérébrale était en faveur d’une leucodystrophie. Le dosage de l’activité de l’aryl sulfatase était effondré à 0 Nm/h/mg. L’enfant a été mis sous valproate de sodium, phénobarbital, et clobazam puis sous topiramate. L’évolution avec un recul de 10 ans a été marquée par des hospitalisations pour des états de mal épileptiques apyrétiques et pour des pneumopathies à répétition.
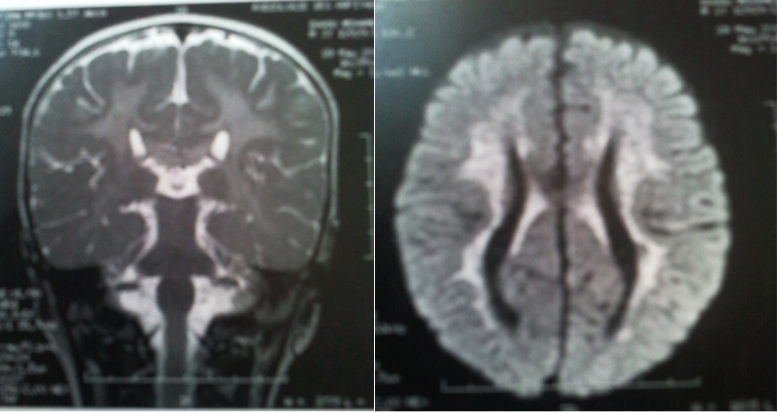
L’enfant M., âgé de 3 ans et demi, consulte pour une régression psychomotrice. L’enfant était issu d’un mariage consanguin (consanguinité de 1er degré), et il avait un bon développement psychomoteur jusqu’à l’âge de 2 ans, quand les parents ont constaté un trouble de la marche et du langage avec une lourdeur des mains. L’examen clinique trouvait une hypertonie de type pyramidal, et l’examen ophtalmologique était normal. La TDM cérébrale avait objectivé une agénésie complète du corps calleux responsable d’un écartement des ventricules latéraux et cornes frontales du ventricule latéral collabées avec un élargissement des citernes de la base et des sillons corticaux frontaux. L’électroencéphalogramme était normal. L’IRM cérébrale avait noté une anomalie de signal de la substance blanche du centre ovale et semi-ovale, un aspect tigré en hypersignal FLAIR avec une restriction de la diffusion, ainsi qu’un hypersignal des bras postérieurs des capsules internes avec respect des fibres en U. L’IRM avait objectivé aussi une anomalie du corps calleux faite d’hypoplasie du corps et du splénium avec une anomalie de signal du genou et du tiers antérieur du corps (figure 4). Le dosage de l’activité de l’aryl sulfatase avait mis en évidence un déficit de l’aryl sulfatase à 0 Nm/h/mg. Un traitement anti épileptique à base de clobazam a été prescrit.
La leucodystrophie métachromatique (LDM) est une maladie de surcharge lysosomale, découverte par Scholz Greenfield en 1925 [3]. Il s’agit d’une maladie rare, due à un déficit en arylsulfatase A (ARSA) [3], enzyme métabolisant une classe de sphingolipides appelés sulfatides. Elle hydrolyse la liaison du sulfate avec le galactose présent dans le sulfatide et dans d’autres glycolipides sulfatés [4]. Le succès de l’hydrolyse de ces sphingolipides par l’ARSA dépend obligatoirement de la présence de la saposine B qui forme un complexe avec le substrat [3]. Le déficit en ARSA conduit à une accumulation de ces sulfatides dans les cellules du système nerveux central (SNC) (myéline, neurones et cellules gliales), entraînant une démyélinisation étendue, resposable d’une atteinte neurologique, un retard mental, des troubles nerveux, et une cécité. Une démyélinisation de nerfs périphériques étant également observée [4].
En ultrastructure, la surcharge cellulaire apparaît sous trois formes :
- – des inclusions lamellaires organisées en « rayon de miel » lorsqu’elles sont coupées transversalement ;
- – des inclusions lamellaires organisées concentriquement et radialement ;
- – des inclusions lamellaires organisées en double membrane, voire en zebra bodies.
Dans les neurones, la surcharge peut se présenter sous la forme de corps cytoplasmiques membranaires. La surcharge implique, dans le système nerveux central, les neurones de structures caudales et les macrophages dans la substance blanche démyélinisée. Au niveau du système nerveux périphérique, la démyélinisation est segmentaire avec présence d’inclusions métachromatiques dans les cellules de Schwann et les macrophages. Ces inclusions métachromatiques peuvent être observées dans d’autres tissus (rein, le pancréas, le foie, la vésicule biliaire, les structures de l’œil) [5]. Le tableau clinique varie selon l’âge de début, ainsi, on distingue trois formes cliniques : la forme infantile (de l’âge d’un à deux ans), la forme juvénile (de 3 à 16 ans) et la forme de l’adulte [5].
La forme infantile est la plus fréquente et la plus sévère. Elle survient après un développement psychomoteur normal dans la première année. La régression motrice procède par étapes, atteignant successivement la position debout, la position assise et la tenue de la tête. À la période initiale, trois tableaux cliniques peuvent être rencontrés
- – celui d’une paralysie flasque avec hypotonie et abolition des réflexes ostéotendineux ;
- – plus fréquemment une combinaison de signes pyramidaux avec abolition des réflexes ostéotendineux ;
- – enfin une paraplégie spastique avec des réflexes vifs.
L’association de signes neurogènes périphériques à des signes pyramidaux évoque fortement le diagnostic. À un stade avancé, on observe une rigidité de décérébration ou des postures de décortication avec des spasmes toniques, périodiques, favorisés par des stimulations et des signes pseudobulbaires. Les fonctions intellectuelles restent longtemps conservées et les crises d’épilepsie sont exceptionnelles. La mort survient entre 3 et 7 ans. Cette forme a été représentée par les observations 1, 2 et 4. La forme juvénile est caractérisée par un arrêt des progrès intellectuels et un effondrement des performances scolaires. Les crises convulsives sont volontiers inaugurales et les signes pyramidaux apparaissent plus tardivement. L’évolution est ralentie par rapport à la forme infantile et le décès survient dans les années qui suivent. Cette forme a été représentée par l’observation 3. La forme de l’adulte est dominée par les signes psychiatriques.
Récemment, Biffi et al, ont proposé une classification des formes cliniques de la LDM en se basant sur le génotype, l’activité enzymatique résiduelle et l’étude de l’expression des protéines mutées [6]. La ponction lombaire met en évidence une hyperprotéinorachie isolée dans les formes infantiles et juvéniles. Le profil électrophorétique du LCR est normal. À l’imagerie, la TDM cérébrale montre des images d’hypodensité symétriques et diffuses au niveau de la SB des deux hémisphères cérébraux et cérébelleux [7]. L’IRM met en évidence une démyélinisation diffuse bilatérale et souvent symétrique, apparaissant en hyper signal en T2 au niveau de la SB péri ventriculaire, sans atteinte des fibres en U. Des images particulières dites en « tigroid » et en « leopard skin » de démyélinisation peuvent se voir dans le centre semi-ovale le corps calleux, la capsule interne, la SB du cervelet et en péri ventriculaire. À des stades plus avancés, une atrophie cortico-sous corticale peut s’observer [8]. Ceci correspond à l’observation 4 a mis en évidence une anomalie de signal de la substance blanche du centre ovale et semi-ovale. La spectroscopie localisée cérébrale de résonance magnétique montre une augmentation du pic de choline intracérébrale, témoin d’une démyélinisation. Le pic de myo-inositol est particulièrement élevé.
L’EMG met en évidence une diminution de la vitesse de conduction nerveuse dans toutes les variétés. Malheureusement, et par faute de moyens, l’EMG a été fait uniquement pour un seul enfant, correspondant à l’observation 1. Le déficit enzymatique en ASA, objectivé dans trois observations [1] [3] [4], reste le diagnostic de confirmation. Il peut être détecté dans les leucocytes du sang ou sur une culture de fibroblastes [9] provenant d’une biopsie de peau. L’ASA est plus effondrée dans les formes infantiles que dans les formes plus tardives. Comme il existe des pseudodéficiences liées à un polymorphisme de l’ASA, assez fréquent dans la population, il est nécessaire de mettre en évidence la surcharge en sulfatides, mise en évidence par la recherche de la sulfatidurie qui doit être un test de première intention. Si elle est présente, et si l’ASA est normale, il faut rechercher un déficit en activateur SAP (sphingolipid activator protein). Ces cas ont été particulièrement observés dans les formes infantiles. Le pseudodéficit en ASA, a été objectivé dans l’observation 2, mais malheureusement, l’enfant est décédé avant de compléter le bilan enzymatique. La biopsie de nerf périphérique confirme le diagnostic, en mettant en évidence la démyélinisation et la présence du matériel métachromatique au niveau des cellules de Schwann et des macrophages [10]. Sur le plan génétique, le gène de l’ASA a été localisé sur le chromosome 22 (22q), où plus de 80 mutations ont été décrites [11]. L’étude du gène qui code pour l’ARSA, est considérée comme le moyen de diagnostic le plus simple et le plus fiable de la maladie, permettant d’éviter les erreurs diagnostiques dues à la présence du pseudodéficit [3]. L’allèle de la pseudodéficience de l’ASA peut être facilement identifié par la biologie moléculaire et de diagnostic facile. Toutefois, les mutations les plus fréquentes dans la population générale sont : la mutation IVS2 + 1G > A associée à la forme infantile tardive, la mutation P426L est fréquemment associée à la forme juvénile, la mutation I179S associée à la forme adulte et les deuxpseudodéficits: N350S et 1524 + 95 A→G (poly A-) [3].
Le diagnostic prénatal reste le meilleur moyen posé chez un couple hétérozygote à la maladie, notamment chez un couple ayant déjà eu un enfant atteint (cas index). Ce diagnostic est possible à partir des cultures de cellules amniotiques ou à partir de la biopsie des villosités choriales par la mesure de l’activité de l’ASA [12] [13]. Il existe trois variantes des LDM :
- -La pseudodéficience en arylsulfatase A pose un problème de diagnostic car elle est présente chez 10% de la population générale [11].
- -La LDM avec déficit en activateur SapB est due à une mutation au niveau du gène SAP (sphingolipid activator protein) identifiée sur le chromosome 10. Ce gène code pour une protéine (sap A B C D) qui stimule l’hydrolyse des sulfatides par l’ASA [11].
- -La maladie d’Austin ou le déficit en multiple sulfatase est une maladie autosomique récessive associant les signes cliniques de LDM et ceux d’une mucopolysaccharidose [14].
Actuellement, aucun traitement ne peut empêcher l’évolution de cette maladie. Les soins palliatifs qui sont donnés visent à soulager la douleur et à améliorer certains symptômes de la maladie, mais n’en arrêtent pas la progression. Sur le plan thérapeutique, la transplantation de la moelle osseuse, effectuée à un stade précoce, avant l’apparition des premiers symptomes, a montré des résultats encourageant dans la forme juvénile de la maladie [15]. La greffe de moelle osseuse (GMO) permet de reconstituer le système hématopoïétique d'un malade grâce à des cellules-souches d'un donneur sain et immunocompatible. Plusieurs malades atteints de forme infantile et juvénile de la LDM ont reçu une greffe de moelle osseuse.
Malgré l'obtention de concentrations comparables à celles des donneurs, aucune régression des signes n'a été rapportée à l'exception d'un enfant qui a eu une amélioration neurologique post-GMO. Par contre, la GMO chez les patients de forme adulte de LDM était très bénéfique. Une amélioration des symptômes neurologiques associée à une normalisation de leurs activités ARSA et leurs taux de sulfatides a été notée. La survie post-GMO chez les patients atteints de la forme adulte est estimée à 77 % [16]. La thérapie génique consiste à introduire dans l'organisme du malade la version normale du gène défectueux responsable de sa maladie [17]. L’AAVrh.10cuARSA est un médicament de thérapie génique, utilisée dans un essai clinique ayant débuté en septembre 2012 en Ile de France. Il s’agit d’un vecteur dérivé du virus AAV, Adeno Associated Virus, modifié non réplicatif contenant le gène humain de l’ARSA. Les premiers résultats de l’essai de thérapie génique pour la leucodystrophie métachromatique (MLD) conduit par le Dr Alessandra Biffi et le Pr Luigi Naldini de l’Institut TIGET San Raffaele à Milan (Italie) viennent d’être publiés. Trois enfants ont été traités et leur maladie a été stoppée. Trois ans après leur traitement, ils vont bien. Cette thérapie offre pour la première fois un espoir de traitement pour les patients atteints de MLD [18].
Les cellules hématopoïétiques modifiées rejoignent la circulation et atteignent le cerveau. Ayant été conçues pour produire en excès la protéine défaillante (l’arylsulfatase A), celles-ci sécrètent dans le milieu extracellulaire la dite protéine, qui est alors captée par les cellules du cerveau alentour. Ainsi, elles métabolisent le sulfate de cérébroside et l'empêchent donc de s'accumuler, empêchant alors la neurodégénérescence de progresser. La thérapie par réduction de substrat par la warfarine, anticoagulant et antagoniste de la vitamine K, a montré que ce composé empêche la synthèse des sphingolipides réduisant de ce fait les concentrations des sulfatides chez les souris. Ainsi la warfarine pourrait être un moyen de diminuer la charge métabolique résultant du défaut de l’ARSA [19]. La thérapie enzymatique consiste à administrer au malade l’enzyme active dont le déficit est responsable de sa maladie. Cette enzyme est administrée au niveau du liquide céphalo-rachidien. Récemment au Danemark, un essai d’enzymothérapie substitutive a été proposé à des enfants présentant la forme infantile tardive. L’enzyme utilisée dans l’essai est semblable à l’enzyme humaine [20].
La leucodystrophie métachromatique (LDM) est une sphingolipidose qui correspond à une perte de myéline dans des régions normalement myélinisées auparavant. Elle est résultante d'un déficit en arylsulfatase A lysosomale. Cette enzyme joue un rôle important dans la cascade enzymatique qui permet le catabolisme des sulfatides [3]. Cette pathologie soulève deux grands problèmes représentés par l’absence d’un traitement efficace, et l’évolution fatale dans la plupart des cas. Actuellement, aucun traitement ne peut empêcher l’évolution de cette maladie. Les soins palliatifs qui sont donnés visent à soulager la douleur et à améliorer certains symptômes de la maladie, mais n’en arrêtent pas la progression. Grace à l’avènement de la thérapie génique, un nouvel espoir de traitement a été offert pour les malades atteints de leucodystrophie métachromatique, mais malheureusement, ce traitement n’est pas généralisé dans tous les pays dont le Maroc en fait partie.
- Heim P, Claussen M, Hoffmann B, Conzelmann E, Gartner J, Harzer K, et al. Leukodystrophy incidence in Germany. Am J Med Genet. 1997;71:475-8 pubmed
- Ozkara H, Topcu M. Sphingolipidoses in Turkey. Brain Dev. 2004;26:363-6 pubmed
- I. Barboura et al. La leucodystrophie métachromatique : Aspects clinique, biologique et thérapeutique. Ann Biol Clin 2010 ; 68 (4) : 385-91.
- Leucodystrophie metachromatique. Disponible à partir du: www.hautconseildesbiotechnologies.fr/IMG/pdf/120625_Leucodystrophie_metachromatique_Avis_CS_HCB-2.pdf
- Powers J, Rubio A. Selected leukodystrophies. Semin Pediatr Neurol. 1995;2:200-10 pubmed
- Jayakumar P, Aroor S, Jha R, Arya B. Computed tomography (CT) in late infantile metachromatic leucodystrophy. Acta Neurol Scand. 1989;79:23-6 pubmed
- Cheon J, Kim I, Hwang Y, Kim K, Wang K, Cho B, et al. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics. 2002;22:461-76 pubmed
- Porter M, Fluharty A, Kihara H. Metachromatic leukodystrophy: arylsulfatase-A deficiency in skin fibroblast cultures. Proc Natl Acad Sci U S A. 1969;62:887-91 pubmed
- Bindu P, Mahadevan A, Taly A, Christopher R, Gayathri N, Shankar S. Peripheral neuropathy in metachromatic leucodystrophy. A study of 40 cases from south India. J Neurol Neurosurg Psychiatry. 2005;76:1698-701 pubmed
- Kaye E. Update on genetic disorders affecting white matter. Pediatr Neurol. 2001;24:11-24 pubmed
- Eto Y, Tahara T, Koda N, Yamaguchi S. Prenatal diagnosis of metachromatic leukodystrophy: a diagnosis with amniotic fluid by DEAE-Sepharose column chromatography and its confirmation by kidney lipid analysis. J Inherit Metab Dis. 1982;5:77-8 pubmed
- Poenaru L, Castelnau L, Besançon A, Nicolesco H, Akli S, Theophil D. First trimester prenatal diagnosis of metachromatic leukodystrophy on chorionic villi by 'immunoprecipitation-electrophoresis'. J Inherit Metab Dis. 1988;11:123-30 pubmed
- Turpin J.-C., Gray F. et Baumann N. Leucodystrophies Editions Techniques-Ency.Méd.Chir. (Paris-France) Neurologie 1994 ; 17-076-D-10, 16p.
- Kidd D, Nelson J, Jones F, Dusoir H, Wallace I, McKinstry S, et al. Long-term stabilization after bone marrow transplantation in juvenile metachromatic leukodystrophy. Arch Neurol. 1998;55:98-9 pubmed
- Krivit W, Saphiro E, Peters C, Lockman LA, Charnas L, Loes D, et al. Adult metachromatic leukodystrophy treated by bone marrow transplantation in 18 patients. SSIEM 39th Annual Symposium Pargue, Czech Republic, 4-7 september 2001. J Inheri Dis 2001 ; 24 : 103.
- Consiglio A, Quattrini A, Martino S, Bensadoun J, Dolcetta D, Trojani A, et al. In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: correction of neuropathology and protection against learning impairments in affected mice. Nat Med. 2001;7:310-6 pubmed
- Crowther M, Ageno W, Garcia D, Wang L, Witt D, Clark N, et al. Oral vitamin K versus placebo to correct excessive anticoagulation in patients receiving warfarin: a randomized trial. Ann Intern Med. 2009;150:293-300 pubmed
- Dali C, Lund AM. thérapie enzymatique substitutive par intraveineuse pour la leukodystrophie metachromatique (MLD). Congrès “Annual Clinical Genetics Meeting”, Tampa, Floride (USA), 25-29 mars 2009.