- A Dibi #asmaadibi at gmail dot comService de Pédiatrie IV- Hôpital d’Enfants de Rabat- CHU Ibn Sina. Faculté de médecine et de Pharmacie – Université Mohamed V Souissi. Rabat-Maroc
- F JabourikService de Pédiatrie IV- Hôpital d’Enfants de Rabat- CHU Ibn Sina. Faculté de médecine et de Pharmacie – Université Mohamed V Souissi. Rabat-Maroc
- A BentahilaService de Pédiatrie IV- Hôpital d’Enfants de Rabat- CHU Ibn Sina. Faculté de médecine et de Pharmacie – Université Mohamed V Souissi. Rabat-Maroc
Introduction : La maladie de Caroli est rare, caractérisée par une dilatation congénitale des voies biliaires, qui peut être localisée ou diffuse. Nous rapportons deux observations pédiatriques de cette maladie.
Observation 1 : Il s’agissait d’une fille âgée de 4 ans, admise à l’hôpital pour ictère cholestatique fébrile avec douleurs abdominales diffuses. A l’examen clinique, il existait une hépatomégalie, et une splénomégalie. La CRP était à 118 mg/l. L’échographie abdominale et le scanner ont mis en évidence une angiocholite sur maladie de Caroli. La patiente a été mise sous antibiothérapie avec une bonne évolution.
Observation 2 : Il s’agissait d’une fille âgée de 13 mois, admise pour distension abdominale évoluant depuis 3 mois, avec pâleur et fièvre. A l’examen clinique, il y avait une hépatomégalie, une circulation veineuse collatérale et une ascite. L’échographie et le scanner ont mis en évidence une maladie de caroli avec angiocholite. La patiente a reçu une antibiothérapie avec une bonne évolution malgré la persistance de l’hépatomégalie.
Conclusion : La maladie de Caroli est une pathologie exceptionnelle chez l’enfant. Cependant, elle doit être évoquée devant des abcès ou des angiocholites à répétition en présence de dilatation des voies biliaires. La transplantation hépatique reste à développer pour traiter les enfants avec forme diffuse.
Introduction: Caroli's disease is rare and characterized by a congenital bile duct dilatation which could be either localized or diffuse. We report two pediatric cases of this disease.
Case report 1: A 4-year-old girl was admitted for febrile cholestatic jaundice with diffuse abdominal pain. Physical examination found hepatomegaly and splenomegaly. CRP was 118 mg/l. Abdominal ultrasound and CT scan were in favor of cholangitis with Caroli syndrome. The patient had antibiotics treatment with good outcome.
Case report 2: A 13-month-old girl was admitted for abdominal distension lasting for three months, with pallor and fever. The examination on admission found hepatomegaly, collateral venous circulation and ascites. Ultrasound and CT scan were in favor of Caroli's disease with cholangitis. The patient received antibiotics with good outcome despite the persistence of hepatomegaly.
Conclusion: Caroli's disease is an exceptional disease in children. However it should be suspected in recurrent cholangitis in the presence of bile duct dilatation. Liver transplantation remains to be developed to treat children with the diffuse form.
La maladie de Caroli est rare, caractérisée par une dilatation congénitale des voies biliaires. Elle peut être localisée ou diffuse. Quand elle est associée à la fibrose hépatique, on parle alors de syndrome de Caroli. Nous rapportons dans ce travail deux observations pédiatriques de maladie de Caroli.

Il s’agissait d’une fille âgée de 4 ans, ayant une notion de consanguinité de 1er degré. Elle a été admise pour un ictère cholestatique fébrile avec des selles décolorées et douleurs abdominales diffuses. L’examen a trouvé des conjonctives normalement colorées, un ictère cutanéo-muqueux, une fièvre à 39,5 °C, un poids à 16 Kg, et une taille à 100 cm. L’examen abdominal a trouvé une cicatrice d’intervention chirurgicale avec un ombilic éversé, une distension abdominale, une hépatomégalie indolore, de consistance ferme, lisse, avec un bord régulier et une flèche hépatique de 13 cm, une splénomégalie à 5 cm au dessous du rebord costal. Le bilan biologique a montré sur la numération formule sanguine (NFS) une anémie normo-chrome normocytaire avec une thrombopénie : hémoglobine (Hb) à 10,8 g/dl, volume globulaire moyen (VGM) à 84µ3, concentration corpusculaire moyenne en hémoglobine (CCMH) à 35,5%, et taux de plaquettes (Plq) à 125000/mm3. Le taux de globules blancs (GB) était normal à 6670/mm3, les polynucléaires (PNN) à 4440/mm3, et les lymphocytes (Lym) à 1710/mm3 (25%). La protéine C réactive (CRP) a été élevée à 118 mg/l. L’ionogramme, la fonction rénale et la protidémie ont été sans particularité. Le bilan hépatique a été perturbé avec des transaminases modérément élevées : alanine amino-transférase (ASAT) à 88 UI/l, aspartate amino-transférase (ALAT) à 76 UI/l), phosphatases alcalines élevées à 536 UI/l, gamma-glutamyl transferase (GGT) élevée à 271 U/l, bilirubine totale élevée à 75 mg/l, bilirubine directe à 52 mg/l, bilirubine indirecte à 23 mg/l. Le reste du bilan hépatique était normal (albuminémie à 31 g/l, cholestérol à 1.42 g/l, triglycérides à 0.74 g/l, taux de prothrombine (TP) à 85%, temps de céphaline activée (TCA) à 30/30). Les sérologies des hépatites virales A, B et C étaient négatives. L’échographie abdominale a montré un foie légèrement augmenté de volume, siège de plusieurs formations échogènes, bien limitées à contenu liquidien impur avec un niveau horizontal, au niveau du segment II de 29 mm de diamètre et du segment III de 31 mm de diamètre, avec présence d’une formation sous-capsulaire hépatique hétérogène, réalisant une empreinte sur les segments VI et VII, et mesurant 64 x 32 mm, en faveur d’un empyème. Par ailleurs, il existait un épaississement péri-portal et une splénomégalie homogène mesurant 110 mm de diamètre longitudinal. Le scanner abdominal a mis en évidence un foie dysmorphique de contours polylobés, avec de multiples formations kystiques bien limitées, de tailles différentes, et de densité identique à celle du liquide biliaire (Figure 1), et dont certains présentent un rehaussement punctiforme intra-lésionnel (dot sign) (Figure 2). Ces formations kystiques étaient essentiellement en périphérie et prédominaient au niveau du segment VI et à cheval des segments II et III, absence de visualisation de la vésicule biliaire, absence de dilatation des voies biliaires extra hépatiques et une splénomégalie homogène mesurant 145 mm de grand axe. Le diagnostic d’angiocholite sur un syndrome de Caroli a été retenu et la patiente a été mise sous antibiothérapie avec une bonne évolution clinique et biologique. Après 2 ans de suivi, la patiente a toujours une hépatomégalie avec le même aspect échographique et la cholestase biologique.

Il s’agit d’une fille âgée de 13 mois, consanguinité de 1er degré, sans antécédent particulier, qui a consulté pour une augmentation du volume abdominal évoluant depuis 3 mois, avec une pâleur, une fièvre non chiffrée. L’examen à l’admission a trouvé un enfant pâle hypotonique, fébrile à 38.5°, poids : 9kg, taille : 79cm, un abdomen distendu avec ombilic déplissé, une hépatomégalie, une circulation veineuse collatérale. La NFS a montré une anémie profonde avec une thrombopénie (Hb : 4.9g/dl, VGM : 80.9 µ3, CCMH : 29.20%, Plq : 50000/mm3, GB : 12700/mm3, PNN : 7340/mm3, Lym : 4390/mm3). L’ionogramme, la fonction rénale et la protidémie ont été sans particularité. Le bilan hépatique était normal en dehors d’une hypoalbuminémie (ASAT : 49UI/l, ALAT : 25UI/l, Alb : 23.58g/l, Cholestérol : 0.74g/l, TG : 1.27g/l, PAL : 186UI/l, Bilirubine Totale : 12mg/l et TP : 71%). La CRP : 30.4mg/l. Le myélogramme était normal. L’échographie abdominale mettait en évidence un foie augmenté de taille, d’échostructure homogène et de contours réguliers avec une vésicule biliaire alithiasique à paroi fine, présence d’une dilatation diffuse des voies biliaires intrahépatique avec des zones ectasiques par endroit, branchées aux voies biliaires, la plus volumineuse de ces ectasies siège au segment VII et mesure 20 mm de diamètre , splénomégalie homogène mesurant 100 mm. Les reins de grande taille, d’echostructure échogène, sans dilatation des cavités excrétrices, avec présence d’un épanchement liquidien intra péritonéal de moyenne abondance. Le scanner abdominal a montré le même aspect en faveur d’une maladie de Caroli diffuse avec des signes d’angiocholite et d’hypertension portale. La fibroscopie œsophagienne a montré l’existence de varices de grade II. La patiente a été mise sous antibiothérapie (ceftriaxone et aminoside) pendant 10 jours avec bonne évolution : disparition de la fièvre, diminution de la CRP, mais persistance de l’hépatomégalie avec même aspect échographique, sans aggravation du bilan hépatique après 9 mois.
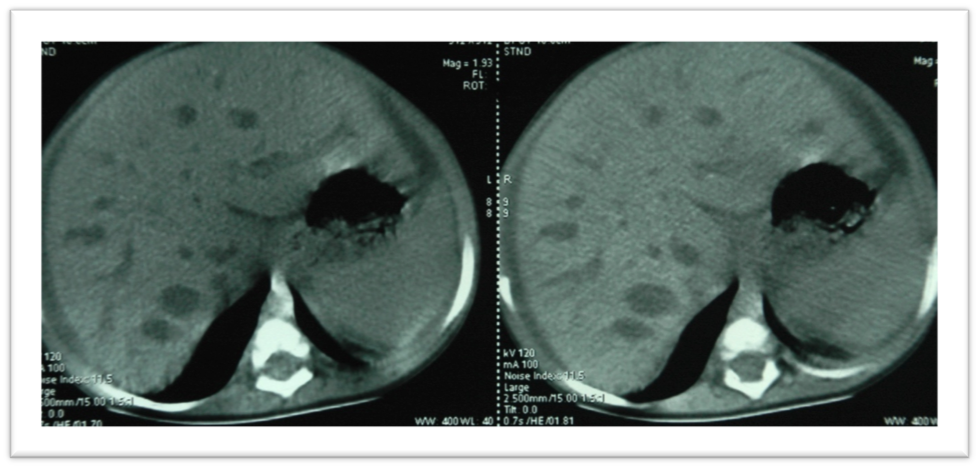
Décrite en 1958 par Caroli et Couinaud [1], la maladie de Caroli est une pathologie congénitale rare qui se définit par une dilatation segmentaire des voies biliaires intra hépatiques diffuse ou localisée dans 20 % des cas (au segment gauche dans 92 % des cas). Elle correspond au type V de la classification des malformations congénitales kystiques des voies biliaires de Todani et al. [2] et se transmet sur le mode autosomique récessif. La prévalence de la maladie est de 1/ 1000000 avec un sex-ratio de 1 [3].
La pathogénie implique une malformation de la plaque ductale pendant le développement embryonnaire [4]. Lorsque le processus intéresse les petits canaux biliaires inter lobulaires, le patient peut développer une fibrose hépatique congénitale [5] qui est la cause de l'hypertension portale dans 20% à 50% des patients avec maladie de Caroli et on parle alors de syndrome de Caroli. Plus de 70% des enfants avec maladie de Caroli présente une fibrose hépatique congénitale, comme le cas de notre malade chez qui la biopsie hépatique a confirmé la présence de la fibrose. Quand la maladie est présente dans un mode familial ou en association avec la fibrose hépatique congénitale, elle est souvent associée à la polykystose rénale [6].
Sur le plan clinique, bien que la maladie de Caroli est présente depuis la naissance, elle reste longtemps asymptomatique et se révèle avant l’âge de 30 ans dans 80 % des cas [7]. Elle se présente sous la forme d’ictère avec fièvre et douleur comme dans nos observations. En effet, l’évolution classique de cette pathologie est dominée par la formation de lithiases intra hépatiques responsables d’angiocholites, d’abcès hépatiques, voire à plus long terme de cholangiocarcinome avec une incidence de 2,5 à 16 % des cas [8].
Le diagnostic positif est confirmé par les examens morphologiques au premier rang desquels se trouve la cholangio-IRM. Ces séquences mettent aisément en évidence les dilatations kystiques des voies biliaires intrahépatiques prédominant autour du hile hépatique et la communication de ces malformations avec le reste de l’arbre biliaire. Le diagnostic peut encore être conforté par la détection d’un flux vasculaire portal ou artériel au sein des malformations kystiques, correspondant au signe central du point (Dot sign) initialement décrit par Choi et al [9] et retrouvé également dans l’observation 1.
Une fois le diagnostic positif de la maladie est posé, l’indication d’un traitement chirurgical radical s’impose afin de prévenir le risque de dégénérescence maligne. Les formes localisées peuvent être traitées par des résections segmentaires ou des hémi hépatectomies tandis que les formes diffuses peuvent justifier une transplantation hépatique [10]. La présence d’autres maladies hépatiques, rendent plus difficile le choix du traitement. Chez nos deux malades, la présence d’une forme diffuse impose une transplantation hépatique. En fait, Bien que peu de publication ont été dédiées au cas pédiatriques de la maladie de Caroli ayant reçus une transplantation hépatique, elle reste à considérer dans les formes diffuses et en présence de complications comme la cirrhose et l’infection. Le plus jeune cas, publié récemment, a été âgé de 7 mois avec une bonne évolution après 57 mois de suivi [11].
La maladie de Caroli est une pathologie exceptionnelle chez l’enfant cependant, elle doit être évoquée devant des abcès ou angiocholite à répétition en présence de dilatation des voies biliaires. La transplantation hépatique reste à développer pour traiter les enfants avec la forme diffuse.
- Caroli J, Soupaul R, Kossakowski J. La dilatation polykystique congénitale des voies biliaires intrahépatiques. Sem. Hop. Paris 1958, 34 : 488 - 95.
- Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134:263-9 pubmed
- Jarry J, Leblanc F, Saric J. Maladie de Caroli monolobaire Presse Med. 2010; 39: 847–848.
- Pinto R, Lima J, da Silveira T, Scholl J, de Mello E, Silva G. Caroli's disease: report of 10 cases in children and adolescents in southern Brazil. J Pediatr Surg. 1998;33:1531-5 pubmed
- Housset C. [Cystic liver diseases. Genetics and cell biology]. Gastroenterol Clin Biol. 2005;29:861-9 pubmed
- Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol. 2003;14:76-89 pubmed
- Dagher I, Franco D. [Cystic diseases of the liver and biliary tract (except for hydatid cyst). Role of surgery]. Gastroenterol Clin Biol. 2005;29:875-7 pubmed
- Mannai S, Kraiem T, Gharbi L, Haoues N, Mestiri H, Khalfallah M. [Congenital cystic dilatation of bile ducts]. Ann Chir. 2006;131:369-74 pubmed
- Luciani A, Kobeiter H, Zegai B, Anglade M, Deux J, Malhaire C, et al. [Imaging in congenital fibrocystic diseases of the liver]. Gastroenterol Clin Biol. 2005;29:870-4 pubmed