Introduction : Les Lymphomes T sous-cutanés de type panniculite (LTSCP) constituent une entité rare de lymphomes, caractérisés par un infiltrat lobulaire de cellules T néoplasiques cytotoxiques CD8+ dont le TCR est de phénotype αβ. Observation : Nous rapportons le cas d'un jeune patient qui a présenté une panniculite associée à une altération de l’état général et chez qui le diagnostic de LTSCP a été posé sur biopsie cutanée. Il a bénéficié d'une polychimiothérapie. Conclusion : L’évolution des LTSCP est chronique et leur pronostic est bon sauf en cas de syndrome hémophagocytaire.
Introduction: Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) are rare. They are characterized by a lobular infiltrate of CD8 + cytotoxic neoplastic T-cells with an α/β+ T-cell phenotype. Case: We report a case of a young patient who presented with panniculitis associated to a poor general condition and who has diagnosed with SPTCL on cutaneous biopsy. He received polychemotherapy. Conclusion: The progression of SPTCL is chronic with a good prognosis except in the case of haemophagocytic syndrome.
Les lymphomes primitifs cutanés représentent un groupe hétérogène de tumeurs de par leur présentation clinique et leur pronostic. Le lymphome T sous-cutané de type panniculite (LTSCP) est une entité à part parmi les lymphomes T périphériques, d’évolution lente et indolente. Son diagnostic demeure difficile et pose le problème de diagnostic différentiel avec d’autres lymphomes T de présentation proche, mais au pronostic plus redoutable, et avec les autres causes de panniculite, en particulier le lupus profond.

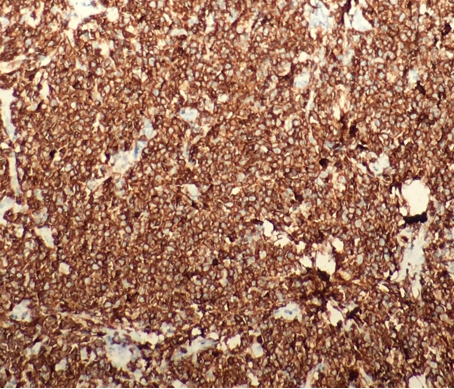
Un homme de 32 ans, sans antécédent particulier, a été hospitalisé devant l’apparition d’une lésion érythémateuse de la racine de la cuisse gauche, d’extension rapide, accompagnée de poussées fébriles chiffrées à 41°C. A l’admission, la lésion située au niveau de la face interne de la cuisse, comprenait une zone inflammatoire de 7 cm de longueur pour une largeur de 4 cm avec une zone indurée douloureuse de 4 cm de grand axe. Il n’existait pas d’ulcération cutanée, d’hypoesthésie, d’impotence fonctionnelle à la marche, d’adénopathie, ou de trouble articulaire. Sur le plan biologique, une bicytopénie était retrouvée, avec 2290 leucocytes/mm3 et une thrombopénie modérée à 148000/mm3, sans anémie (hémoglobinémie à 14,8 g/dL). La protéine C réactive était élevée (46 mg/L). De plus, le complément du bilan biologique objectivait une augmentation modérée des triglycérides plasmatiques (2,19 mmol/L), une hyperferritinémie sérique (20200 µg/L), une augmentation de la bilirubine totale et conjuguée plasmatique à 26 et 17 µmol/L, un taux de CPK plasmatique à 164 mg/L et un taux plasmatique de LDH à 3983 UI/L. Le scanner de la racine des membres inférieurs, abdominal et thoracique retrouvait une infiltration de la graisse sous-cutanée de la cuisse gauche sans abcès, ainsi que des plans sous-cutanés et de la graisse mésentérique en regard de l’orifice inguinal droit. Il n’existait pas d’adénopathie abdomino-pelvienne, médiastinale, ou d’hépatosplénomégalie. Après huit jours d’hospitalisation, la persistance des signes cliniques (avec une fièvre en plateau autour de 40°C) et biologiques, malgré une antibiothérapie par voie intra-veineuse associant oxacilline, gentamicine et métronidazole, a conduit rapidement à la réalisation d’une biopsie cutanée profonde. Elle a permis d’observer une infiltration cellulaire de densité modérée intéressant l’hypoderme et le derme profond, dissociant les adipocytes, et respectant le derme superficiel et l’épiderme. Elle était constituée de nombreux macrophages comprenant une hémophagocytose, et surtout une prolifération de cellules rondes de nature lymphoïde, de taille petite à moyenne, aux noyaux irréguliers et hyperchromatiques (figures 1 et 2). Ces cellules lymphoïdes exprimaient les marqueurs T, ainsi que CD8, sans expression de CD4, de CD56 et de CD30 (figures 3 et 4). Ces aspects morphologiques, corroborés au profil immunohistochimique, permettaient de proposer le diagnostic de lymphome T sous-cutané de type panniculite.
Dans les suites immédiates, on observait une diffusion des lésions cutanées au niveau des flancs et du thorax, l’exacerbation du syndrome d’activation macrophagique, mais l’absence d’envahissement ostéo-médullaire, conduisant à proposer devant cette forme disséminée, une polychimiothérapie (de type ACVB).

Les lymphomes T de l’hypoderme sont rares et de d’identification relativement récente, décrite initialement par Gonzalez en 1991 [1]. Actuellement, le lymphome T sous-cutané à type de panniculite, a été reconnu comme une entité à part parmi les autres de lymphomes à cellules T périphériques [2, 3]. Ils représentent moins de 1 % des lymphomes non-hodgkiniens. Ils s’observent à tout âge, mais atteignent préférentiellement l’adulte jeune autour de la quatrième décennie, avec un sex-ratio proche de 1 [4].
Cliniquement, il se présente sous forme de plaques disséminées ou de multiples nodules sous-cutanés érythémateux, de taille variée, parfois hémorragiques, voire ulcérés. Les lésions se localisant préférentiellement au niveau des extrémités, en particulier sur les membres inférieurs et le tronc. Il n’existe en principe pas d’atteinte ganglionnaire, sauf dans les stades disséminés tardifs. Un syndrome d’activation macrophagique (SAM) est associé dans près de la moitié des cas, pouvant se manifester par une altération de l’état général (avec fièvre, sueurs et asthénie) et une hépatosplénomégalie, des adénopathies périphériques, une éruption cutanée morbilliforme, voire des signes de défaillance multiviscérale dans les formes fulminantes [1]. Les anomalies biologiques sont nombreuses, non spécifiques, souvent majeures, conduisant à évoquer le diagnostic de SAM devant leur association (bi- ou pancytopénie, troubles de l’hémostase, voire une réelle CIVD, une cytolyse ou une insuffisance hépatique.
Le diagnostic du LTSCP est anatomopathologique, souvent difficile, nécessitant des prélèvements biopsiques profonds. Sur le plan histologique, l’atteinte plus ou moins massive du tissu sous-cutanée est évocatrice, dissociant les lobules graisseux. Une infiltration moins dense, avec des cellules disséminées entre les lobules graisseux et réalisant un aspect « en dentelle » doit également attirer l’attention. L’hypoderme est envahi par une population lymphoïde atypique, pléomorphe, constituée de cellules lymphomateuses de taille petite à grande, aux noyaux irréguliers, parfois masquée par l’infiltration macrophagique, associée à des phénomènes d’hémophagocytose. Des aspects de cytophagie (débris nucléaires intra-macrophagiques) ou de caryorrhexie sont constants, avec en principe, absence de nécrose adipocytaire [5]. L’analyse immunohistochimique montre que les cellules lymphomateuses expriment CD2, CD3, CD7, CD8, mais pas CD4, avec un profil cytotoxique (expression habituelle de TIA-1, de la Perforine et du Granzyme B), sans expression de CD56. Dans les cas difficiles, le recours à la biologie moléculaire met en évidence un clone T α/β. Ces caractéristiques permettent d’éliminer le lymphome T sous-cutané, s’étendant souvent au derme, de type γ/, au profil immunohistochimique CD4-, CD8-, CD56+, et de plus mauvais pronostic [2, 3]. Ce profil permet également d’éliminer les autres types de lymphome T pouvant s’accompagner d’une localisation sous-cutanée, non exclusive ou marginale [6] : le lymphome « natural killer » (NK) blastique ; le lymphome T/NK « de type nasal » cutané, caractérisé par la prolifération de lymphocytes CD56+, exprimant CD30 de façon hétérogène et le virus Epstein Barr; le lymphome anaplasique; et enfin le mycosis fongoïde et le lymphome T épidermotrope CD8+. Toutefois le principal diagnostic à éliminer en dehors d’une pathologie lymphomateuse est la panniculite lupique ou lupus profond, pour laquelle la distinction peut apparaître impossible, si bien que la relation entre ces deux entités reste controversée d’autant que leur présentation clinique peut être similaire et que le LTSCP accompagné d’un SAM peut être associé à des maladies dysimmunitaires dans près de 20 % des cas [4, 5].

L’évolution de ce type de lymphome T est particulière puisqu’il est rattaché à une faible évolutivité, avec des cas de régression spontanée, sauf en présence d’un SAM [2, 4, 6]. Globalement leur pronostic est excellent, avec des taux de survie à 5 ans de l’ordre de 82 à 91 % [6].
Ces considérations cliniques et évolutives incitent généralement à une certaine retenue en matière thérapeutique, pouvant aller jusqu’à l’abstention thérapeutique dans les formes indolentes n’échappant pas à un suivi clinique régulier et attentif. Dans les formes chroniques, la corticothérapie orale peut suffire à contrôler de la maladie, avec comme alternative, en cas de mauvaise tolérance, la radiothérapie locale, la monochimiothérapie à base de méthotrexate ou de ciclosporine [4]. La polychimiothérapie (CHOP, ESHAP) est réservée aux formes graves disséminées, accompagnées d’un SAM, comme dans le cas rapporté.
Les LTSCP sont des lymphomes rares, caractérisés par un infiltrat lobulaire de cellules T néoplasiques cytotoxiques CD8+ dont le TCR est de phénotype αβ. L’évolution est chronique et leur pronostic est bon sauf en cas de syndrome hémophagocytaire.
Au stade initial, le tableau clinique de panniculite spontanément résolutive ou corticosensible associé à un infi ltrat lobulaire lymphohistiocytaire aux atypies cellulaires peu marquées rendent le diagnostic de LTSP diffi cile.
Le suivi attentif des patients présentant un tableau de panniculite, la répétition des biopsies cutanées profondes, l’analyse complète clinique, évolutive, histologique, immunophénotypique et moléculaire est indispensable pour affirmer le diagnostic de LTSP et le différencier des panniculites dites « bénignes ».
Il n’existe aucun conflit d’intérêts concernant les données publiées dans cet article entre les auteurs.
Tous les auteurs ont contribué à l'élaboration de ce travail sur toutes ses étapes depuis l'acquisition et l'exploitation des données, jusqu'à la rédaction et la révision et enfin l'approbation finale de la version de cet article. Tous les auteurs ont contribué à la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
- Willemze R, Jaffe E, Burg G, Cerroni L, Berti E, Swerdlow S, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-85 pubmed
- Delsol G. The 2008 WHO lymphoma classification. Ann Pathol. 2008 Nov; 28 Spec No 1(1):S20-4.
- Willemze R, Jansen P, Cerroni L, Berti E, Santucci M, Assaf C, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111:838-45 pubmed
- Takeshita M, Imayama S, Oshiro Y, Kurihara K, Okamoto S, Matsuki Y, et al. Clinicopathologic analysis of 22 cases of subcutaneous panniculitis-like CD56- or CD56+ lymphoma and review of 44 other reported cases. Am J Clin Pathol. 2004;121:408-16 pubmed